Acetyl-CoA is a principal substrate feeding TCA. cycle and energy production. Brain displays high demand for energy due to high frequency of neuronal depolarizatio-repolarization cycles.. Therefore, adequate provision of acetyl-CoA precursors is critical factor for proper neuronal activity and survival.
- acetyl-CoA metabolism
- neurodegenerative diseases
- zinc dyshomeostasis
- thiamine deficiency
1. Introduction
1. Introduction
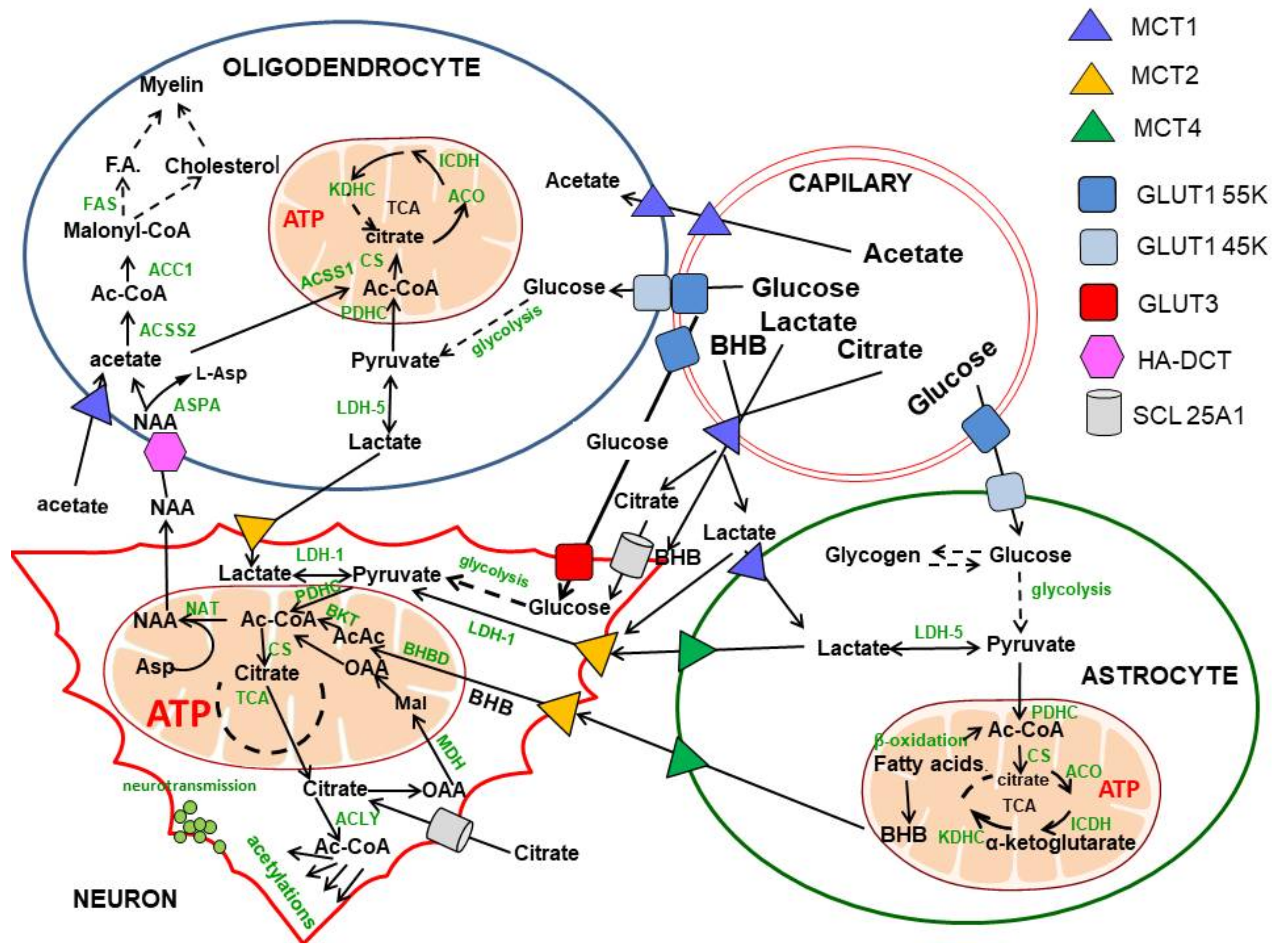
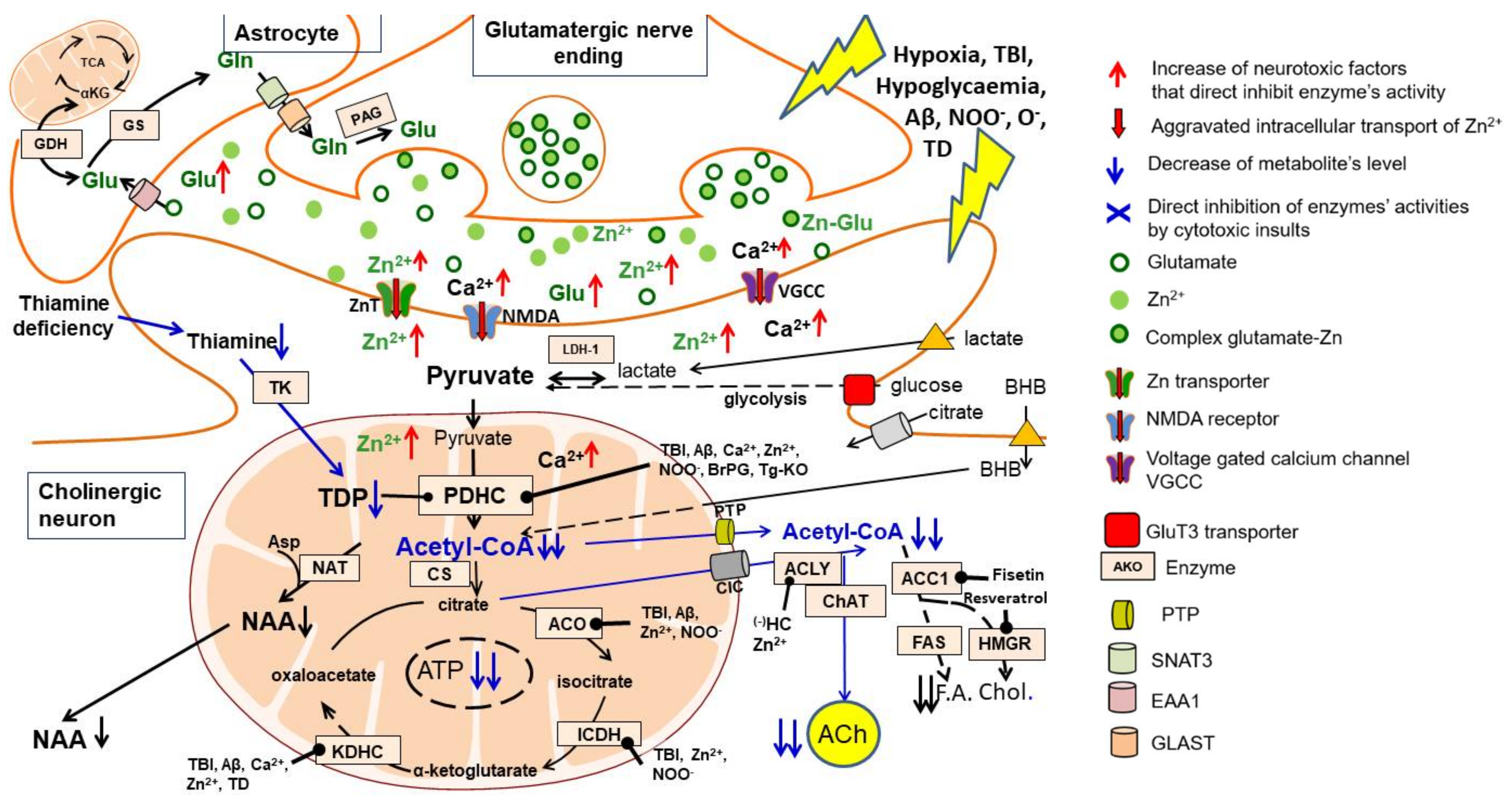
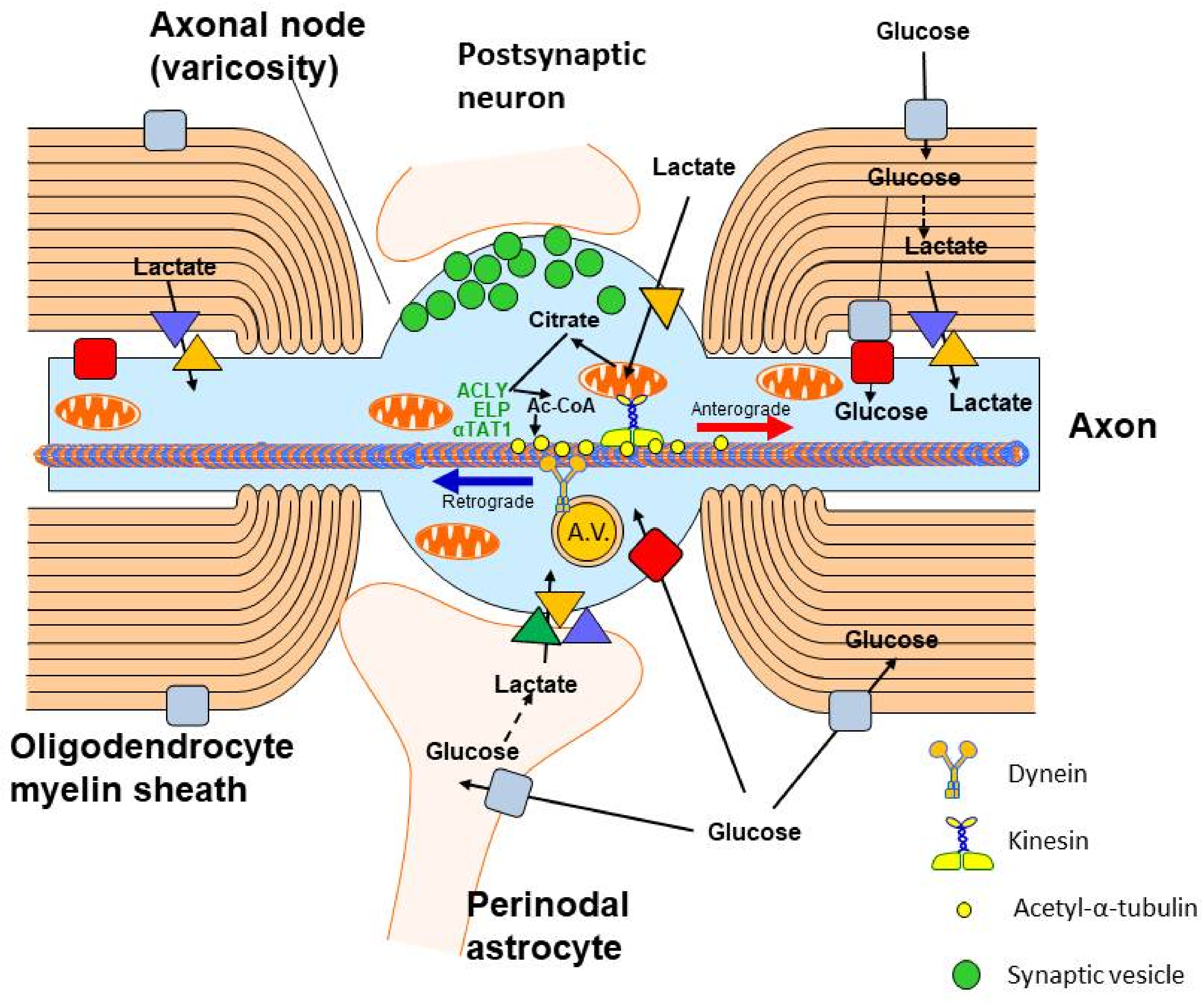
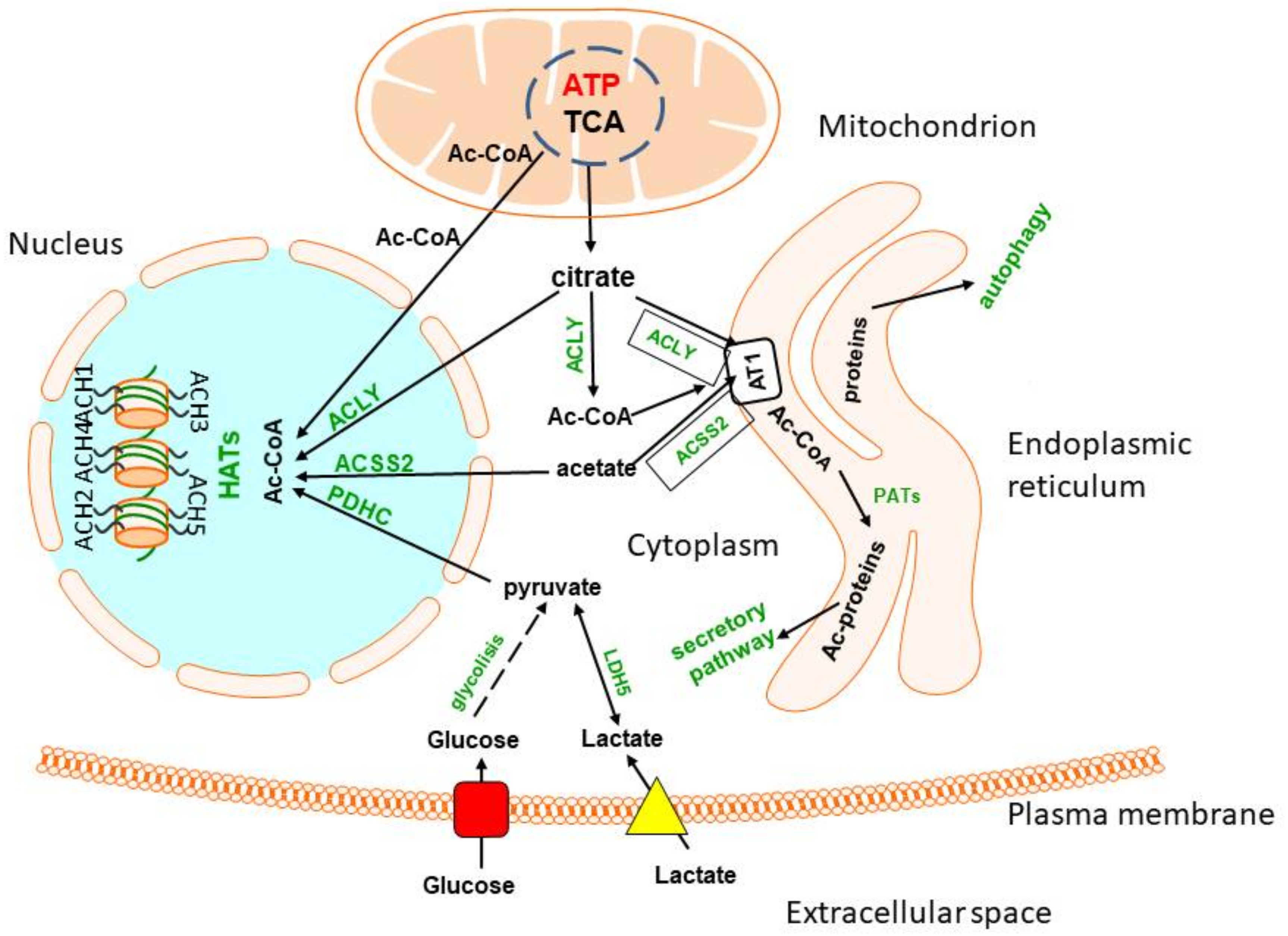
2. Glucose and Lactate—The Key Precursors of Brain Acetyl-CoA in Health and Disease
Table 1. Levels of acetyl-CoA in different brain compartments in various experimental models of brain pathologies.
References
- Belanger, M.J.; Allman, I.; Magistretti, P.J. Brain energy metabolism. Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.C.; Dienel, C.A.; Sonnewald, H.S.; Waagepetersen, H.S.; Schousboe, A. Energy metabolism of the brain. In Basic Neurochemistry: Principles of Molecular, Cellular and Medical Neurobiology, 8th ed.; Brady, S., Siegel, G., Albers, R.W., Donald, L., Price, D.L., Eds.; Elsevier B.V.: Amsterdam, The Netherlands, 2012; pp. 200–231. [Google Scholar]
- Divakaruni, A.S.; Wallace, M.; Buren, C.; Martyniuk, K.; Andreyev, A.Y.; Li, E.; Fields, J.A.; Cordes, T.; Reynolds, I.J.; Bloodgood, B.L.; et al. Inhibition of the mitochondrial pyruvate carrier protects from excitotoxic neuronal death. J. Cell Biol. 2017, 216, 1091–1105. [Google Scholar] [CrossRef] [PubMed]
- Beard, E.; Lengacher, S.; Dias, S.; Magistretti, P.J.; Finsterwald, C.H. Astrocytes as key regulators of brain energy metabolism: New therapeutic perspectives. Front. Physiol. 2022, 12, 825816. [Google Scholar] [CrossRef] [PubMed]
- Bonvento, G.; Bolaños, J.P. Astrocyte-neuron metabolic cooperation shapes brain activity. Cell Metab. 2021, 33, 1546–1564. [Google Scholar] [CrossRef]
- Zhang, S.; Lachance, B.; Mattson, M.P.; Jia, X. Glucose metabolic crosstalk and regulation in brain function and diseases. Prog. Neurobiol. 2021, 204, 102089. [Google Scholar] [CrossRef]
- Sun, W.; Cornwell, A.; Li, J.; Peng, S.; Osorio, J.; Aslling, N.; Wang, S.; Benraiss, A.; Lou, N.; Goldman, S.A.; et al. S0X9 is an astrocyte-specific nuclear marker in the adult brain outside the neurogenic regions. J. Neurosci. 2017, 37, 4493–4507. [Google Scholar] [CrossRef]
- Popov, A.; Branze, N.; Fedotova, A.; Tiaglik, A.; Bychkov, M.; Morozova, N.; Branze, A.; Aronov, D.; Lyukmanova, E.; Lazareva, N.; et al. A high-fat diet changes astrocytic metabolism to promote synaptic plasticity and behavior. Acta Physiol. 2022, 236, e13847. [Google Scholar] [CrossRef]
- Bhatt, D.P.; Rosenberger, T.A. Acetate treatment increases fatty acid content in LPS-stimulated BV2 microglia. Lipids 2014, 49, 621–631. [Google Scholar] [CrossRef]
- Currais, A.; Huang, L.; Petrascheck, M.; Maher, P.; Schubert, D. A chemical biology approach to identifying molecular pathways associated with aging. Geroscience 2021, 43, 353–365. [Google Scholar] [CrossRef]
- Zyśk, M.; Bielarczyk, H.; Gul-Hinc, S.; Dyś, A.; Gapys, S.; Ronowska, A.; Sakowicz-Burkiewicz, M.; Szutowicz, A. Phenotype-dependent interaction between N-acetyl-L-aspartate and acetyl CoA in septal SN56 cholinergic cells exposed to an excess of zinc. J. Alzheimer Dis. 2017, 56, 1145–1158. [Google Scholar] [CrossRef]
- Janssen, L.; Ai, X.; Zheng, X.; Wei, W.; Caglayan, A.B.; Kilic, E.; Wang, Y.-C.; Hermann, D.M.; Venkataramani, V.; Bähr, M.; et al. Inhibition of fatty acid synthesis aggravates brain injury, reduces blood-brain barrier integrity and impairs neurological ecoverry in a murine stroke model. Front. Cell. Neurosci. 2021, 15, 327. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [PubMed]
- Ronowska, A.; Szutowicz, A.; Bielarczyk, H.; Gul-Hic, S.; Klimaszewska-Łata, J.; Dyś, A.; Zyśk, M.; Jankowska-Kulawy, A. The regulatory effects of acetyl-CoA distribution in the healthy and diseased brain. Front. Cell. Neurosci. 2018, 12, 169–189. [Google Scholar] [CrossRef]
- Szutowicz, A.; Bielarczyk, H.; Zyśk, M.; Dyś, A.; Ronowska, A.; Gul-Hinc, S.; Klimaszewska-Łata, J. Early and late pathomechanisms in Alzheimer’s disease: From zinc to amyloid-β neurotoxicity. Neurochem. Res. 2017, 42, 891–904. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, P.C. Acetyl-CoA metabolism and histone acetylation in the regulation of aging and lifespan. Antioxidants 2021, 10, 572. [Google Scholar] [CrossRef]
- Schuberth, J.; Sollenberg, J.; Sundvall, A.; Sörbo, B. Acetylcoenzyme A in brain. J. Neurochem. 1966, 13, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Ičny, J.; Tuček, S. Acetyl-coenzyme and acetylcholine in slices of rat caudate nuclei incubated in the presence of metabolic inhibitors. J. Biol. Chem. 1981, 256, 4919–4923. [Google Scholar]
- Bielarczyk, H.; Szutowicz, A. Evidence for the regulatory function of synaptoplasmic acetyl-CoA in acetylcholine synthesis in nerve endings. Biochem. J. 1989, 262, 337–380. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the cholinergic system. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef]
- Simpson, I.A.; Carruthers, A.; Vannucci, S.J. Supply and demand in cerebral energy metabolism: The role of nutrient transporters. J. Cereb. Blood Flow Metab. 2007, 27, 1766–1791. [Google Scholar] [CrossRef]
- Szablewski, L. Glucose transporters in brain: In health and in Alzheimer’s disease. J. Alzheimers Dis. 2017, 55, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.V.; Crumpacker, R.H.; Calderon, K.E.; Garcia, F.G.; Zbesko, J.C.; Frye, J.B.; Gonzalez, S.; Becktel, D.A.; Yang, T.; Tavera-Garcia, M.A.; et al. Post-stroke administration of the p75 neurotrophin receptor modulator, LM11A-31, attenuates chronic changes in brain metabolism, increases neurotransmitter levels, and improves recovery. J. Pharmacol. Exp. Ther. 2022, 380, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Patching, S.G. Glucose transporters at the blood-brain barrier: Function, regulation and gateways for drug delivery. Mol. Neurobiol. 2017, 54, 1046–1077. [Google Scholar] [CrossRef]
- Sharma, V.; Singh, T.G. Therapeutic implications of glucose transporters (GLUT) in cerebral ischemia. Neurochem. Res. 2022, 47, 2173–2186. [Google Scholar] [PubMed]
- Pérez-Escuredo, J.; Van Hée, V.F.; Sboarina, M.; Falces, J.; Payen, V.L.; Pellerin, L.; Sonveaux, P. Monocarboxylate transporters in the brain and in cancer. Biochim. Biophys. Acta 2016, 1863, 2481–2497. [Google Scholar] [CrossRef] [PubMed]
- Roosterman, D.; Cottrell, G.S. Astrocytes and neurons communicate via a monocarboxylic acid shuttle. AIMS Neurosci. 2020, 7, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I.A. cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef]
- Wohnsland, S.; Bürgers, H.F.; Kuschinsky, W.; Maurer, M.H. Neurons and neuronal stem cells survive in glucose-free lactate and in high glucose cell culture medium during normoxia and anoxia. Neurochem. Res. 2010, 35, 1635–1642. [Google Scholar] [CrossRef]
- Li, Y.; Lu, B.; Sheng, L.; Zhu, Z.; Sun, H.; Zhou, Y.; Yang, Y.; Xue, D.; Chen, W.; Tian, X.; et al. Heksokinase 2-dependent hyperglycolisis driving microglial activation contributes to ischemic brain injury. J. Neurochem. 2018, 144, 186–200. [Google Scholar] [CrossRef]
- Sutendra, G.; Kinnaird, A.; Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Hashimoto, K.; Zhang, N.; Flaim, E.; Michelakis, E.D. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 2014, 158, 84–97. [Google Scholar] [CrossRef]
- Li, X.; Zhang, J.; Li, D.; He, C.H.; He, K.; Xue, T.; Wan, L.; Zhang, C.H.; Liu, Q.; Wan, L.; et al. Astrocytic ApoE reprograms neuronal cholesterol metabolism and histone-acetylation-mediated memory. Neuron 2021, 109, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Zhao, T.; Du, J.; Ji, G.; Li, X.; Ji, S.; Tian, W.; Wang, X.; Hao, A. TIGAR promotes neural stem cell differentiation through acetyl-CoA-mediated histone acetylation. Cell Death Dis. 2019, 10, 198. [Google Scholar] [CrossRef]
- Sun, Y.; Li, T.; Xie, C.; Zhang, Y.; Zhou, K.; Wang, X.; Blomgren, K.; Zhu, C.H. Dichloroacetate treatment improves mitochondrial metabolism and reduces brain injury in neonatal mice. Oncotarget 2016, 7, 31708–31722. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Bian, X.; Zhang, Q.; Xia, Z.; Liu, B.; Chn, Q.; Ke, C.; Wu, J.L.; Zhao, Y. Shengui sansheng san ameliorates cerebral energy deficiency via citrate cycle after ischemic stroke. Front. Pharmacol. 2019, 10, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Khoury, N.; Xu, J.; Stegelman, S.D.; Jackson, C.W.; Koronowski, K.B.; Dave, K.R.; Young, J.I.; Perez-Pinzon, M.A. Resveratrol reconditioning induces genomic and metabolic adaptations with long-term window of cerebral ischemic tolerance leading to bioenergetic efficiency. Mol. Neurobiol. 2019, 56, 4549–4565. [Google Scholar] [CrossRef] [PubMed]
- Koronowski, K.B.; Khoury, N.; Saul, N.; Loriz, Z.B.; Cohan, C.H.; Stradecki-Cohan, H.M.; Dave, K.R.; Young, J.I. Neuronal SIRT1 (silent information regulator 2 homologue 1) regulates glycolysis and mediates resveratrol-induced ischemic tolerance. Stroke 2017, 48, 3117–3125. [Google Scholar] [CrossRef]
- Klimaszewska-Łata, J.; Gul-Hinc, S.; Bielarczyk, H.; Ronowska, A.; Zyśk, M.; Grużewska, K.; Pawełczyk, T.; Szutowicz, A. Differential effects of lipopolysaccharide on energy metabolism in murine microglial N9 and cholinergic SN56 neuronal cells. J. Neurochem. 2015, 133, 284–297. [Google Scholar] [CrossRef]
- Gul-Hinc, S.; Michno, A.; Zyśk, M.; Szutowicz, A.; Jankowska-Kulawy, A.; Ronowska, A. Protection of cholinergic neurons against zinc toxicity by glial cells in thiamine-deficient media. Int. J. Mol. Sci. 2021, 22, 13337–13357. [Google Scholar] [CrossRef]
- Ronowska, A.; Gul-Hinc, S.; Bielarczyk, H.; Pawełczyk, T.; Szutowicz, A. Effects of zinc on SN56 cholinergic neuroblastoma cells. J. Neurochem. 2007, 103, 972–983. [Google Scholar] [CrossRef]
- Greco, T.; Vespa, P.M.; Prins, M.L. Alternative substrate metabolism depends on cerebral metabolic state following traumatic brain injury. Exp. Neurol. 2020, 329, 113289. [Google Scholar] [CrossRef]
- Narayanan, S.E.; Rehuman, N.A.; Harilal, S.; Vincent, A.; Rajamma, R.G.; Behl, T.; Uddin, M.S.; Ashraf, G.M.; Mathew, B. Molecular mechanism of zinc neurotoxicity in Alzheimer’s disease. Environ. Sci. Pollut. Res. Int. 2020, 35, 43542–43552. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Levenson, C.W. Zinc and traumatic brain injury: From chelation to supplementation. Med. Sci. 2020, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Ronowska, A.; Gul-Hinc, S.; Michno, A.; Bizon-Zygmańska, D.; Zyśk, M.; Bielarczyk, H.; Szutowicz, A.; Gapys, B.; Jankowska-Kulawy, A. Aggravated effects of coexisting marginal thiamine deficits and zinc excess on SN56 neuronal cells. Nutr. Neurosci. 2021, 6, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Annoni, F.; Peluso, L.; Bogossian, E.G.; Creteur, J.; Zanier, E.R.; Taccone, F.S. Brain protection after anoxic brain injury: Is lactate supplementation helpful ? Cells 2021, 10, 1714. [Google Scholar] [CrossRef] [PubMed]
- Duhaut, D.E.; Heurteaux, C.; Gandin, C.; Ichai, C.; Quintard, H. The antiedematous effect of exogenous lactate therapy in traumatic brain injury: A physiological and mechanistic approach. Neurocrit. Care 2021, 35, 747–755. [Google Scholar] [CrossRef]
- Wang, P.; Chen, M.; Yang, Z.; Yu, T.; Zhu, J.; Zhou, L.; Lin, J.; Fang, X.; Huang, Z.; Jiang, L.; et al. Activation of pyruvate dehydrogenase activity by dichloroacetate improves survival and neurologic outcomes after cardiac arrest in rats. Shock 2018, 49, 704–711. [Google Scholar] [CrossRef]
- Szutowicz, A.; Bielarczyk, H.; Skulimowska, H. Effect of dichloroacetate on acetyl-CoA content and acetylcholine synthesis in rat brain synaptosomes. Neurochem. Res. 1994, 19, 1107–1112. [Google Scholar] [CrossRef]
- Glancy, B.; Balaban, R.S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef]
- Piao, L.; Fang, Y.H.; Kubler, M.M.; Donnino, M.W.; Sharp, W.W. Enhanced pyruvate dehydrogenase activity improves cardiac outcomes in a murine model of cardiac arrest. PLoS ONE 2017, 12, e0185046. [Google Scholar] [CrossRef]
- Ikeda, K.; Liu, X.; Kida, K.; Marutani, E.; Hirai, S.; Sakaguchi, M.; Andersen, L.W.; Bagchi, A.; Cocchi, M.N.; Berg, K.M.; et al. Thiamine as a neuroprotective agent after cardiac arrest. Resuscitation 2016, 105, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Kho, A.R.; Choi, B.Y.; Lee, S.H.; Hong, D.K.; Jeong, J.H.; Kang, B.S.; Kang, D.H.; Park, K.H.; Park, J.B.; Suh, S.W. The effects of sodium dichloroacetate on mitochondrial dysfunction and neuronal death following hypoglycemia-induced injury. Cells 2019, 8, 405. [Google Scholar] [CrossRef] [PubMed]
- Jakkamsetti, V.; Marin-Valencia, I.; Ma, Q.; Good, L.B.; Terrill, T.; Rajasekaran, K.; Pichumani, K.; Khemtong, C.H.; Hooshyar, M.A.; Sundarrajan, C.H.; et al. Brain metabolism modulates neuronal excitability in a mouse model of pyruvate dehydrogenase deficiency. Sci. Transl. Med. 2019, 11, eaan0457. [Google Scholar] [CrossRef] [PubMed]
- Jakkamsetti, V.; Ma, Q.; Pascual, J.M. A subset of synaptic transmission events is coupled to acetyl coenzyme A production. J. Neurophysiol. 2022, 127, 623–636. [Google Scholar] [CrossRef]
- Chevalier, A.C.; Rosenberger, T.A. Increasing acetyl-CoA metabolism attenuates injury and alters spinal cord lipid content in mice subjected to experimental autoimmune encephalomyelitis. J. Neurochem. 2017, 141, 721–737. [Google Scholar] [CrossRef]
- Della-Flora Nunes, G.; Mueller, L.; Silvestri, N.; Patel, M.S.; Wrabetz, L.; Feltri, M.L.; Poitelon, Y. Acetyl-CoA production from pyruvate is not necessary for preservation of myelin. Glia 2017, 65, 1626–1639. [Google Scholar] [CrossRef]
- Lazzarino, G.; Amorini, A.M.; Signoretti, S.; Musumeci, G.; Lazzarino, G.; Caruso, G.; Pastore, F.S. Pyruvate dehydrogenase and tricarboxylic acid cycle enzymes are sensitive targets of traumatic brain injury induced metabolic derangement. Int. J. Mol. Sci. 2019, 20, 5774. [Google Scholar] [CrossRef]
- Bubber, P.; Haroutunian, V.; Blass, J.P.; Gibson, G.E. Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann. Neurol. 2005, 57, 695–703. [Google Scholar] [CrossRef]
- Huang, Z.; Yan, Q.; Wang, Y.; Zou, Q.; Li, J.; Liu, Z.; Cai, Z. Role of mitochondrial dysfunction in the pathology of amyloid-β. J. Alzheimers Dis. 2020, 78, 505–514. [Google Scholar] [CrossRef]
- Hoshi, M.; Takashima, A.; Murayama, M.; Yoshida, N.; Hohino, T. Nontoxic amyloid beta peptide 1–42 suppresses acetylcholine synthesis. Possible role in cholinergic dysfunction in Alzheimer’s disease. J. Biol. Chem. 1997, 272, 2038–2041. [Google Scholar]
- Bielarczyk, H.; Jankowska-Kulawy, A.; Höfling, C.; Ronowska, A.; Gul-Hinc, S.; Roβner, S.; Schliebs, R.; Pawełczyk, T.; Szutowicz, A. AβPP-transgenic 2576 mice mimic cell type-specific aspects of acetyl-CoA-linked metabolic deficits in Alzheimer’s disease. J. Alzheimer Dis. 2015, 48, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Gandbhir, O.; Sundaram, P. Effect of AmyTrap, an amyloid-β binding drug, on Aβ induced mitochondrial dysfunction and tau phosphorylation in cultured neuroblastoma cells. Metab. Brain Dis. 2020, 35, 923–931. [Google Scholar] [CrossRef] [PubMed]
| Experimental Model | Signal/Conditions | Acetyl-CoA Level/Relative Change | Reference/Comments |
|---|---|---|---|
| Rat brain | Hypoxia in vivo | Whole tissue (nmol/g tissue) | [17] |
| Control | 5.4 | ||
| Hypoxia 100N2 90 s | 6.7 ** | ||
| Rat brain | Brain region (whole tissue) | Whole tissue (nmol/g tissue) | [65] |
| Thalamus | 9.1 | ||
| Hippocampus | 7.1 | ||
| Cortex | 6.2 | ||
| Cerebellum | 6.1 | ||
| Rat brain slices | 60 min. incubation 31.2 mM K+ | Brain slices (nmol/g tissue) | [18] |
| Control | 5.04 | ||
| +3-bromorypuvate 0.25 mM | 2.45 | ||
| Rat brain synaptosomes | 30 min. incubation 30 mM K+ | Synaptosomes Mitochondria Cytoplasm |
[19] |
| (pmol/mg protein) | |||
| Control | 12.3 46.8 | ||
| +3-bromopyruvate 0.25 mM | 0 7.4 ** | ||
| Healthy adult rat brain synaptosomes | Healthy control | Whole synaptosomes (pmol/mg protein) | [66] different from pyruvate alone, † p < 0.05 |
| Substrate used (mM) | |||
| Pyr. 2.5 BHB 2.5 Pyr. + BHB | |||
| 24.3 7.1 22.8 | |||
| STZ diabetes 10 d | 31.3 * 10.5 * 29.4 * | ||
| Streptozotocin-diabetic rat brain synaptosomes | STZ diabetes 10 d + Insulin 5 d | 30.6 * 10.0 *† 35.6 *† | |
| Cholinergic neuroblastoma cell culture: nondifferentiated (NC) and differentiated (DC, db-cAMP 1 mM + retinoic acid (RA) 0.001 mM 48 h) | Control | Cellular compartment levels (pmol/mg protein) |
[67] from respective NC, † p < 0.05, †† p < 0.01 |
| Mitochondria Cytoplasm | |||
| NC DC NC DC | |||
| 71 22 † 13 50 † | |||
| +NGF 100 ng/mL 24 h | 55 42 ** 71 ** 29 *† | ||
| Native SN56TrkA-/p75NTR+ | Control | 95 23 † 13 49 † | |
| Tg T17 SN56TrkA+/p75NTR+ | +NGF 100 ng/mL 24 h | 59 * 39 * 129 ** 48 † | |
| Cholinergic neuroblastoma cell culture Tg T17 SN56TrkA+/p75NTR+ NC, and DC |
24 h cell culture with: |
Relative change against no addition control (%) |
[68][69] Different from respective NC, † p < 0.05, †† p < 0.01; from Aβ (25–35) alone, ‡ p < 0.05, ‡‡ p < 0.01 |
| Mitochondria Cytoplasm | |||
| NC DC NC DC | |||
| Aβ25-35 0.001 mM | 10 −23 −17 −58 ** | ||
| Acetyl-carnitine 0.1 mM | +39 ** 0 † 0 +54 **†† | ||
| Aβ + acetyl-carnitine | +22 ‡‡ 0 0 0 ‡‡ | ||
| ILβ 10 ng/mL | −11 −18 +38 * −42 *†† | ||
| Aβ + ILβ | −18 −1 +1 +3 ‡‡ | ||
| Cholinergic neuroblastoma cell culture | ChAT (nmol/min/mg protein) NC DC |
Whole cells (pmol/mg protein) NC DC |
[70] Different from respective native SN56, † p < 0.05, †† p < 0.01 |
| Native SN56 TrkA-/p75NTR+ | 0.22 0.79 *** | 31.2 21.9 *** | |
| Tg T17 TrkA+/p75NTR+ | 0.19 0.47 *** | 39.7 † 26.8 ***† | |
| Tg ChAT2 TrkA-/p75NTR+ | 3.80 ††† 6.80 ***††† | 15.5 †† 11.2 *** †† | |
| Cholinergic neuroblastoma cells Native SN56 TrkA-/p75NTR + DC |
24 h cell culture with: Control |
Mitochondria Cytoplasm (pmol/mg protein) |
[40] † different from ZnCl2 0.10 mmol/L |
| 11.8 20.9 | |||
| ZnCl2 0.10 mM | 9.3 19.6 | ||
| ZnCl2 0.15 mM | 11.4 13.5 *† | ||
| Cholinergic neuroblastoma cells Native SN56 TrkA-/p75NTR + NC and DC |
30 min incubation (protein free medium) with: Zn 0.1 mM |
Relative change vs. no Zn control (%) Mitochondria Cytoplasm NC DC NC DC −5 −35 ** −100 ** −80 ** |
[71] |
| Subcutaneous pyrithiamine (PT) 0.025 mg/kg b.w./day and thiamine free diet 14 d Rat forebrain synaptosomes |
PT synaptosomes vs. no PT control | Forebrain synaptosomesRelative change against no PT control (%) | [72] |
| Mitochondria Cytoplasm | |||
| No Ca Ca 1.0mM no Ca Ca1.0mM | |||
| −53 *** −35 *** −43 *** −24 * | |||
| Subcutaneous PT 0.025 mg/kg b.w./day and thiamine-free diet 14 d. Rat forebrain whole mitochondria | PT whole forebrain mitochondria vs. no PT control | Forebrain whole mitochondria Relative change vs. no PT control (%) |
[73] |
| No Ca Ca 0.01 mM ADP/HX | |||
| −62 *** −62 *** −52 *** | |||
| Cholinergic neuroblastoma cell culture Native SN56 TrkA-/p75NTR+ |
Thiamine-free culture medium 48 h +Amprolium 2 mM |
Relative change vs. no amprolium NC control (%) |
[74] Amprolium suppressed TPP level—28% vs. control |
| Mitochondria Cytoplasm. | |||
| NC DC NC DC | |||
| −43 −57 −58 *** −50 ** | |||
| Endoplasmic reticulum from WT and AT 1-1S113R/+ mice | Mutation AT 1-1S113R/+ | Acetyl-CoA transport (pmol/mg/5 min.) | [75] |
| WT 370 | |||
| AT-1S113R/+ 142 *** | |||
| N9 microglioma cells culture | 24 h culture with: LPS 0.01 µg/mL |
Relative change against respective no addition control (%) |
[38] ‡‡ different from SNP 0.4 mM, p < 0.01 ††† different from N9 cells, p < 0.001 |
| Whole cells | |||
| N9 SN56 | |||
| −23 * +4 | |||
| SynchronizedCholinergic neuroblastoma cells Native SN56 TrkA-/p75NTR+ DC |
SNP 0.4 mM | −3 −38 * | |
| LPS + SNP | −6 92 ***†††‡‡ | ||
| WT 14–16 mos mouse brain AβPP-Tg 2576 14-16 m mouse brain | Accumulation about 0.6 μM Aβ1-42 in Tg brain | Relative change vs. WT control (%) Mitochondria Cytoplasm ** |
[62] Acetyl-CoA—control WT mice Synapt. mitoch. 39 pmol/mg prot. Synapt. cytopl. 90 pmol/mg prot. Whole brain mitoch. 45 pmol/mg. |
| Forebrain synaptosomes | −44 ** −34 | ||
| Forebrain whole mitochondria | +5 - | ||
| WT mouse brain AT1 Tg mouse brain (overexpression) |
Hippocampus Isolated adult neurons H4 neuroglioma |
AT1 Tg vs. WT Relative difference (%) Cytoplasm |
[76] |
| −41 * | |||
| −45 * | |||
| −43 * | |||
| WT 9 d postnatal mouse brain | 24 h post hypoxia/ischemia | Relative change vs. control (%) Mitochondrial fraction Vehicle-treated DCA-treated +6 +27 * |
[34] |
| Cell line cultures WT SN56 TrkA-/p75NTR NC |
Intracellular Zn accumulation of 5 nmol/mg protein at extracellular Zn in culture medium: 0.125 mM |
Relative change vs. no Zn control (%) SN56 NC −54 *** |
[11] † different from NC, p < 0.05 |
| DC | 0.110 mM |
SN56 DC −48 ***† | |
| SHSY5Y dopaminergic neurons | 0.150 mM | SHSY5Y −31 * | |
| C6 astroglioma | 0.200 mM | C6 −44 ** | |
| 3XTg AD 16.5 mos mouse brain | 8 mos ketone ester-feeding | Relative change vs. non-ketotic control (%) Hippocampus +79 * |
[77] Acetyl-CoA no ketone control: 17 μmol/g tissue |
| Mouse BV2 microglial cells culture | Dimethylsulfoxide-induced 6 h hypoxia | Relative change vs. no hypoxia control (%) +79 ** |
[30] |
| Hypoxia + Lonidamine 0.05 mM | −58 * | ||
| Hypoxia + 3-Bromopuryvate | −42 * | ||
| Cholinergic neuroblastoma cells WT SN56 TrkA-/p75NTR+ DC |
30 min incubation (protein-free medium) with: Control Nifedipine 0.01 mM GVIA 0.0005 mM MVIIC 0.0002 mM |
Whole cells (pmol/mg protein) No Zn Zn 0.15 mM 30.5 13.8 * 30.7 29.2 † 28.8 21.6 *† 28.1 20.5 *† |
[78] Compounds used here are inhibitors of different types of calcium channels. * p < 0.01 vs. no Zn control; † < 0.01 vs. 0.15 mM Zn. |
| SAMP8 mice brain cortex | 13 mos vs. 9 mos change No treatment Fed with CMS121 4 mos Fed with J147 4 mos |
Relative change 13 mos vs. 9 mos | [79] CMS121, J147 are acetyl-CoA carboxylase inhibitors. |
| (%) | |||
| −41 **** | |||
| −12 | |||
| −6 | |||
HT22 hippocampal neuronal cell culture Primary E21 mice neuronal culture |
24 h culture with: | Relative change vs. no addition control (%) |
[10][79] Compounds used here inhibit acetyl-CoA carboxylase by different mechanisms. |
| +ACC1 siRNA | +114 *** | ||
| +TOFA 0.01 mM | +178 *** | ||
| +CMS 121 0.001 mM | +140 *** | ||
| +J147 0.001 mM | +100 ** | ||
| +CAD031 0.001 mM | +177 *** | ||
| +CMS 121 0.001 mM | +57 *** | ||
| +J147 0.001 mM | +29 | ||
| +CAD031 0.001 mM | +108 *** | ||
| Brain-specific pdha1flox8/wt deficient mice (PDHD) | PDHD |
Relative change vs. control (%) −12 |
[54] |
| 3xTgAD mice WT control mice |
Ageing—2, 11, 21 mos hippocampus whole tissue Control |
2 mos 11 mos 21 mos | [80] Different from the corresponding 2 mos mice, † p < 0.05, ††† p < 0.001 |
| (Arbitrary units) | |||
| Male | |||
| 0.5 1.1† 1.3 ††† | |||
| 3XTgAD | 1.2 * 1.6 * 2.6 **††† | ||
| Female | |||
| Control | 0.5 0.8 1.0 † | ||
| 3XTgAD | 0.5 1.3 *† 1.2 † | ||
| Rat permanent middle cerebral artery occlusion model of brain stroke (pMCAO) | Shengui Shanseng San (SSS) extraction feeding per os 3 d before and 7 d after pMCAO | Relative change vs. sham control | [35] Absolute sham control value of infarct-corresponding control region equal to 24.4 µmol/µL tissue is 106 times higher than those reported elsewhere. |
| In brain infarcted region (%) pMCAO −80 *** |
|||
| Low dose SSS + pMCAO −52 *** | |||
| Middle dose SSS +pMCAO −44 *** | |||
| High dose SSS +pMCAO −4 | |||
| Closed-head impact acceleration model of mild or severe traumatic rat brain injury (mTBI/sTBI) | mTBI/sTBI | Relative change vs. control (%) Whole brain extracts |
[58] Absolute control value about 39 nmol/g wet weight is about 10 times higher than values reported elsewhere. Different from the corresponding of post mTBI time, † p < 0.005 |
| Post mTBI 6 h −13 | |||
| 24 h −22 | |||
| 48 h −24 | |||
| 120 h −13 | |||
| Post sTBI 6 h −34 * | |||
| 24 h −56 *† | |||
| 48 h −47 *† | |||
| 120 h −58 *† | |||
| HEK293 cell culture | DIP2A overexpression |
Relative change DIP2A vs. no insert control (%) +120 * |
[81] |
| Traumatic brain injury/control cortical impact rat brain (TBI/CCI) | TBI/CCI | Peri-contusional brain cortex acetyl-CoA (ng/mg protein) Early immediate 3 h i.v. administration |
[41] Absolute control value is about 34.5 pmol/mg protein. |
| Sham (saline 0.9%) 27 | |||
| Control 38 | |||
| Glucose 30% 57 * | |||
| Lactate 100 mM 29 | |||
| BHB 2M 52 * | |||
| Late (6 h post impact) 3 h i.v. administration | |||
| Glucose 30% 38 | |||
| Lactate 100 mM 21 | |||
| BHB 2M 38 | |||
| Cholinergic neuroblastoma cells WT SN56 TrkA-/p75NTR+ DC |
30 min incubation (protein-free medium) with: | Relative change vs. control (%) Mitochondria Cytoplasm |
[82] Mecamylamine is a nonselective antagonist of nicotinic receptors. 2APB is inhibitor of IP3 receptors and TRP channels. |
| Mecamylamine 0.002 mM | −36 ** +7 | ||
| Nifedipine 0.01 mM | 0 +28 | ||
| 2-Aminoethoxydiphenyl borate (2-APB) 0.05 mM | +43 -56 ** | ||
| Zn 0.15 mM | −64 *** −39 ** | ||
| Human fibroblastoma HT1080 cell line ACLY WT ACLY-WT ACLY KO |
4 h incubation with or without 20 mM acetate ACLY-WT ACLY-KO |
Relative change vs. WT-acetate control (%) | [83] Absolute control value for ACLY-WT is 6.1 μM (normalised to internal standard) |
| acetate 20 mM No acetate | |||
| 0 −14 | |||
| −67 *** −95 *** | |||
| E18 C57BL/6J mice model of AD | 24 h culture with Aβ1-42 10µM |
Relative change vs. control (%) | [84] Absolute control values for neurons and microglia are 0.45 and 0.75 μM, respectively |
| Neurons Microglia ** | |||
| 0 −31 | |||
| 5XFAD 9 mos mouse brain | 5XFAD control 5XFAD + efavirenz 0.1 mg/kg b.w./d in drinking water from 3 to 9 mos of life |
Whole brain Mitochondria (pmols/mg protein) |
[85] Efavirenz is an inhibitor of reverse transcriptase. Acetyl-CoA control levels reported here are about 10 times higher than reported elsewhere. |
| 145 87 | |||
| 351 *** 352*** | |||
| B6SJ/L 9 months mouse brain | B6SJ/L control | 361 157 | |
| Tg Cyp46a1+/+ | Tg Cyp46a1+/+ | 257 *** 128 | |
| Tg Cyp46a1−/− | Tg Cyp46a1−/− | 143 *** 100 *** | |
| Cholinergic neuroblastoma cells WT SN56 TrkA-/p75NTR+ NC and DC |
24 h culture in thiamine-free medium with: +Zn 0.1 mM +Amprolium 5 mmol/L +Zn +Amprolium |
Relative change vs. no Zn, and amprolium control (%) Mitochondria |
[45] Absolute control acetyl-CoA levels in NC and DC mitochondria were: 11.6 and 11.9 pmol/mg protein, respectively. Absolute control acetyl-CoA levels in NC and DC cytoplasm were: 13.6 and 11.7 pmol/mg protein, respectively. †† different from NC/DC Zn, p < 0.0.1 different from NC/DC amprolium, ‡ p < 0.05, ‡‡ p < 0.01 |
| NC DC | |||
| −5 −23 ** | |||
| −5 −16 * | |||
| −45 ***††‡‡ -50 **††‡‡ | |||
| Cytoplasm | |||
| +Zn 0.1 mM | −4 −12 | ||
| +Amprolium 5 mmol/L | −17 −12 | ||
| Thiamine-deficient culture medium | +Zn +Amprolium | −54 **††‡‡ −53 **††‡‡ | |
| C6 astroglioma cells Cholinergic neuroblastoma cells WT SN56 TrkA-/p75NTR+ DC |
24 h culture C6 in thiamine-free or thiamine-supplemented medium with: Amprolium 10 mM Zn 0.15 mM Zn 0.20 mM 24 h culture SN56 in thiamine-free medium in co-culture with C6 C6 co-culture Amprolium 5 mM Zn 0.1 mM Amprolium + Zn Amprolium + Zn+C6 co-culture |
Relative change vs. no Zn, no amprolium control (%) Thiamine deficient Thiamine suppl. |
[39] Absolute control levels of acetyl-CoA in SN56 and C6 cells were: 27.2 and 14.6 pmol/mg protein, respectively. † different from Amprolium+Zn, p < 0.05 |
| −26 ** 0 | |||
| −28 −16 | |||
| −68 ** −56 ** | |||
| Relative change vs. no co-culture, Zn, and no amprolium control (%) | |||
| +10 | |||
| −26 | |||
| −29 | |||
| −64 * | |||
| −10 † | |||
| WT mouse brain C57BL/6J mouse brain |
Glycerol triacetate 3 g/kg b.w./d 10 d by gavage, and euthanised 60 min. post last gavage | Hippocampus Relative change vs. control (%) |
[86] |
| Whole tissue Nuclei Cytoplasm | |||
| +171 * +19 *** +13 ** | |||
| Non-fasted mouse brain | Sacrificed 30 min. post oral ketone esters (KE) administration 3 mg/g b.w | Relative change KE vs. control (%) Brain cortex +114 *** |
[87] |
| Cultured primary neurons (E17 C57BL/6J mice) | Astrocyte-derived ApoE particles Astrocyte-derived medium (ADM)Apo E enriched ADMApo E depleted ADM |
Relative change vs. no ApoE control (%) Acetyl-CoA/CoA ratio Whole cells Nuclei +86 * +175 *** Acetyl-CoA/CoA ratio +200 *** +40 |
[32] |
| WT mouse brain Elp3 conditional KO mouse brain |
Lack Elongator to Atat1 activity |
Relative change vs. WT control (%) Cortical neurons−72 |
[88] |
| WT mouse brain C57Bl/6J mouse brain |
Acute stress |
Relative change vs.no stress control (%) Prefrontal cortex +113 * |
[89] Absolute acetyl-CoA level, 0.37 pmol/μg |
| C57BL/6J mice—stroke and hypoxia | 12 wk post-stroke oral administration p75 NTR modulator (LM11A-31) | Relative change vs. sham control (%) Brain infarcted region None LM11A-31 −32 +36 * |
[23] |
| Primary astrocytes—0–1-day-old mice cerebral cortex U87 human glioblastoma cells U87FABP7wt U87FABPmut. U251human glioma cells U251 FABP7KO |
FABP7-KO vs. WT cells FABP7wt vs. control FABP7mut vs. control FABP7KO vs. control |
Relative change vs. WT control (%) Whole cells Isolated nuclei −34 * −28 * +87 * +74 * −10 −39 −48 * −70 * |
[90][91] Absolute acetyl-CoA for control WT cells is 450 pmol/106, and 74 pmol/107 nuclei. |
| WT mouse brain SLC25A1 nTg mouse brain |
Hippocampus and cortex cytoplasm Lumen of the endoplasmic reticulum |
Relative change vs. WT control (%) Cytoplasm ER +58 *** +72 **** |
[92] SLC25A1 nTg—mitochondrial citrate carrier |
|
Experimental model |
Signal/conditions |
Acetyl-CoA level/ relative change |
Reference/comments |
|
Cholinergic neuroblastoma cells Native SN56 TrkA-/p75NTR+DC
|
24 h cell culture with: Control ZnCl2 0.10 mmol/L ZnCl2 0.15 mmol/L |
Mitochondria Cytoplasm (pmol/mg protein) 11.8 20.9 9.3 19.6 11.4 13.5*† |
[40]
†differrent from ZnCl2 0.10 mmol/L
|
|
N9 microglioma cells culture
Cholinergic neuroblastoma cells Native SN56 TrkA-/p75NTR+ DC |
24h culture with:
LPS 0.01 µg/mL SNP 0.4 mM LPS + SNP |
Relative change against respective no addition control (%) Whole cells N9 SN56 -23* +4 -3 -38* -6 - 92***†††‡‡ |
[38]
‡‡ different from SNP 0.4 mM p<0.01 †††different from N9 cells p<0.001 |
|
WT 9 d postnatal mouse brain |
24 h post hypoxia/ischemia |
Relative change vs. control (%) Mitochondrial fraction Vehicle-treated DCA treated +6 +27* |
[34] |
|
Cell line cultures
WT SN56 TrkA-/p75NTR NC DC
SHSY5Y dopaminergic neuro.
C6 astroglioma |
Intracellular Zn accumulation of 5 nmol/mg protein at extracellular Zn in culture medium:
0.125 mM 0.110 mM
0.150 mM
0.200 mM |
Relative change vs. no Zn control (%)
SN56 NC -54*** SN56 DC -48***†
SHSY5Y -31*
C6 -44** |
[11]
†different from NC, p<0.05
|
|
Mouse BV2 microglial cells culture |
Dimethylsulfoxide induced 6 h hypoxia Hypoxia+Lonidamine 0.05 mM Hypoxia +3-Bromopuryvate |
Relative change vs. no hypoxia control ( %) +79** -58* -42* |
[30]
|
|
Brain-specific pdha1flox8/wt deficient mice (PDHD) |
PDHD |
Relative change vs. control (%) -12 |
[54] |
|
Rat permanent middle cerebral artery occlusion model of brain stroke (pMCAO) |
Shengui Shanseng San (SSS) extraction feeding per os 3 d before and 7 d after pMCAO |
Relative change vs. sham control in brain infarcted region (%) pMCAO -80*** Low dose SSS + pMCAO -52*** Middle dose SSS +pMCAO -44*** High dose SSS + pMCAO -4 |
[35] Absolute sham control value of infarct-corresponding control region equal to 24.4 µmol/µl tissue is 106 times higher than those reported elsewhere. |
|
Closed-head impact acceleration model of mild or severe traumatic rat brain injury (mTBI/sTBI) |
mTBI/sTBI |
Relative change vs. control (%) Whole brain extracts Post mTBI 6 h -13 24 h -22 48 h -24 120 h -13 Post sTBI 6 h -34* 24 h -56*† 48 h -47*† 120 h -58*† |
[58] Absolute control value about 39 nmol/g wet weight is about 10 times higher from values reported elsewhere. different from the corresponding of post mTBI time †p<0.005
|
|
Traumatic brain injury/control cortical impact rat brain (TBI/CCI) |
TBI/CCI |
Peri-contusional brain cortex acetyl-CoA (ng/mg protein) Early immediate 3 h i.v. administration Sham (saline 0.9%) 27 Control 38 Glucose 30% 57* Lactate 100 mM 29 BHB 2M 52* Late (6 h post impact) 3h i.v. administration Glucose 30% 38 Lactate 100 mM 21 BHB 2M 38 |
[41]
Absolute control value is about 34.5 pmol/mg protein.
|
|
C6 astroglioma cells
Cholinergic neuroblastoma cells
WT SN56 TrkA-/p75NTR+ DC
|
24h culture C6 in thiamine-free or thiamine supplemented medium with: Amprolium 10 mM Zn 0.15 mM Zn 0.20 mM
24h culture SN56 in thiamine-free medium in co-culture with C6 C6 co-culture Amprolium 5 mM Zn 0.1 mM Amprolium + Zn Amprolium +Zn+C6 co-culture |
Relative change vs. no Zn, no amprolium control (%) Thiamine deficient Thiamine suppl. -26** 0 -28 -16 -68** -56**
Relative change against no co-culture, Zn, and no amprolium control (%)
+10 -26 -29 -64* -10†
|
[39]
Absolute control levels of acetyl-CoA in SN56 and C6 cells were: 27.2 and 14.6 pmol/mg protein, respectively.
†different from Amprolium+Zn p<0.05 |
The majority of the data is presented as relative (%) change versus respective control value. It results from the fact that they are presented in arbitrary units. In some cases absolute values of acetyl-CoA are given to enable quantitative assessment of this metabolite. Distributions data are deleted for clarity. Significance of differences between groups is marked by superscript symbols. Data significantly different from respective control, * p<0.05; ** p<0.01; *** p<0.001; **** p<0.0005. Other comparisons are given as individual references.
Abbreviations: BHB, β-hydroxubutyrate; mTBI/sTBI, mild/severe traumatic brain injury; PDHD, pyruvate dehydrogenase deficiency; pMCAO, permanent middle cerebral artery occlusion; SSS, Shenggui Sansheng San composed of Panax ginseng root and rhizome, Angelica sinensis root and rhizome, Cinnamomum cassia;.
References
- Belanger, M.J.; Allman, I.; Magistretti, P.J. Brain energy metabolism. Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738.
- McKenna, M.C.; Dienel, C.A.; Sonnewald, H.S.; Waagepetersen, H.S.; Schousboe, A. Energy metabolism of the brain. In Basic Neurochemistry: Principles of Molecular, Cellular and Medical Neurobiology, 8th ed.; Brady, S., Siegel, G., Albers, R.W., Donald, L., Price, D.L., Eds.; Elsevier B.V.: Amsterdam, The Netherlands, 2012; pp. 200–231.
- Divakaruni, A.S.; Wallace, M.; Buren, C.; Martyniuk, K.; Andreyev, A.Y.; Li, E.; Fields, J.A.; Cordes, T.; Reynolds, I.J.; Bloodgood, B.L.; et al. Inhibition of the mitochondrial pyruvate carrier protects from excitotoxic neuronal death. J. Cell Biol. 2017, 216, 1091–1105.
- Beard, E.; Lengacher, S.; Dias, S.; Magistretti, P.J.; Finsterwald, C.H. Astrocytes as key regulators of brain energy metabolism: New therapeutic perspectives. Front. Physiol. 2022, 12, 825816.
- Bonvento, G.; Bolaños, J.P. Astrocyte-neuron metabolic cooperation shapes brain activity. Cell Metab. 2021, 33, 1546–1564.
- Zhang, S.; Lachance, B.; Mattson, M.P.; Jia, X. Glucose metabolic crosstalk and regulation in brain function and diseases. Prog. Neurobiol. 2021, 204, 102089.
- Sun, W.; Cornwell, A.; Li, J.; Peng, S.; Osorio, J.; Aslling, N.; Wang, S.; Benraiss, A.; Lou, N.; Goldman, S.A.; et al. S0X9 is an astrocyte-specific nuclear marker in the adult brain outside the neurogenic regions. J. Neurosci. 2017, 37, 4493–4507.
- Popov, A.; Branze, N.; Fedotova, A.; Tiaglik, A.; Bychkov, M.; Morozova, N.; Branze, A.; Aronov, D.; Lyukmanova, E.; Lazareva, N.; et al. A high-fat diet changes astrocytic metabolism to promote synaptic plasticity and behavior. Acta Physiol. 2022, 236, e13847.
- Bhatt, D.P.; Rosenberger, T.A. Acetate treatment increases fatty acid content in LPS-stimulated BV2 microglia. Lipids 2014, 49, 621–631.
- Currais, A.; Huang, L.; Petrascheck, M.; Maher, P.; Schubert, D. A chemical biology approach to identifying molecular pathways associated with aging. Geroscience 2021, 43, 353–365.
- Zyśk, M.; Bielarczyk, H.; Gul-Hinc, S.; Dyś, A.; Gapys, S.; Ronowska, A.; Sakowicz-Burkiewicz, M.; Szutowicz, A. Phenotype-dependent interaction between N-acetyl-L-aspartate and acetyl CoA in septal SN56 cholinergic cells exposed to an excess of zinc. J. Alzheimer Dis. 2017, 56, 1145–1158.
- Janssen, L.; Ai, X.; Zheng, X.; Wei, W.; Caglayan, A.B.; Kilic, E.; Wang, Y.-C.; Hermann, D.M.; Venkataramani, V.; Bähr, M.; et al. Inhibition of fatty acid synthesis aggravates brain injury, reduces blood-brain barrier integrity and impairs neurological ecoverry in a murine stroke model. Front. Cell. Neurosci. 2021, 15, 327.
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821.
- Ronowska, A.; Szutowicz, A.; Bielarczyk, H.; Gul-Hic, S.; Klimaszewska-Łata, J.; Dyś, A.; Zyśk, M.; Jankowska-Kulawy, A. The regulatory effects of acetyl-CoA distribution in the healthy and diseased brain. Front. Cell. Neurosci. 2018, 12, 169–189.
- Szutowicz, A.; Bielarczyk, H.; Zyśk, M.; Dyś, A.; Ronowska, A.; Gul-Hinc, S.; Klimaszewska-Łata, J. Early and late pathomechanisms in Alzheimer’s disease: From zinc to amyloid-β neurotoxicity. Neurochem. Res. 2017, 42, 891–904.
- Bradshaw, P.C. Acetyl-CoA metabolism and histone acetylation in the regulation of aging and lifespan. Antioxidants 2021, 10, 572.
- Schuberth, J.; Sollenberg, J.; Sundvall, A.; Sörbo, B. Acetylcoenzyme A in brain. J. Neurochem. 1966, 13, 819–822.
- Ičny, J.; Tuček, S. Acetyl-coenzyme and acetylcholine in slices of rat caudate nuclei incubated in the presence of metabolic inhibitors. J. Biol. Chem. 1981, 256, 4919–4923.
- Bielarczyk, H.; Szutowicz, A. Evidence for the regulatory function of synaptoplasmic acetyl-CoA in acetylcholine synthesis in nerve endings. Biochem. J. 1989, 262, 337–380.
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the cholinergic system. Curr. Neuropharmacol. 2016, 14, 101–115.
- Simpson, I.A.; Carruthers, A.; Vannucci, S.J. Supply and demand in cerebral energy metabolism: The role of nutrient transporters. J. Cereb. Blood Flow Metab. 2007, 27, 1766–1791.
- Szablewski, L. Glucose transporters in brain: In health and in Alzheimer’s disease. J. Alzheimers Dis. 2017, 55, 1307–1320.
- Nguyen, T.V.; Crumpacker, R.H.; Calderon, K.E.; Garcia, F.G.; Zbesko, J.C.; Frye, J.B.; Gonzalez, S.; Becktel, D.A.; Yang, T.; Tavera-Garcia, M.A.; et al. Post-stroke administration of the p75 neurotrophin receptor modulator, LM11A-31, attenuates chronic changes in brain metabolism, increases neurotransmitter levels, and improves recovery. J. Pharmacol. Exp. Ther. 2022, 380, 126–141.
- Patching, S.G. Glucose transporters at the blood-brain barrier: Function, regulation and gateways for drug delivery. Mol. Neurobiol. 2017, 54, 1046–1077.
- Sharma, V.; Singh, T.G. Therapeutic implications of glucose transporters (GLUT) in cerebral ischemia. Neurochem. Res. 2022, 47, 2173–2186.
- Pérez-Escuredo, J.; Van Hée, V.F.; Sboarina, M.; Falces, J.; Payen, V.L.; Pellerin, L.; Sonveaux, P. Monocarboxylate transporters in the brain and in cancer. Biochim. Biophys. Acta 2016, 1863, 2481–2497.
- Roosterman, D.; Cottrell, G.S. Astrocytes and neurons communicate via a monocarboxylic acid shuttle. AIMS Neurosci. 2020, 7, 94–106.
- Magistretti, P.J.; Allaman, I.A. cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901.
- Wohnsland, S.; Bürgers, H.F.; Kuschinsky, W.; Maurer, M.H. Neurons and neuronal stem cells survive in glucose-free lactate and in high glucose cell culture medium during normoxia and anoxia. Neurochem. Res. 2010, 35, 1635–1642.
- Li, Y.; Lu, B.; Sheng, L.; Zhu, Z.; Sun, H.; Zhou, Y.; Yang, Y.; Xue, D.; Chen, W.; Tian, X.; et al. Heksokinase 2-dependent hyperglycolisis driving microglial activation contributes to ischemic brain injury. J. Neurochem. 2018, 144, 186–200.
- Sutendra, G.; Kinnaird, A.; Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Hashimoto, K.; Zhang, N.; Flaim, E.; Michelakis, E.D. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 2014, 158, 84–97.
- Li, X.; Zhang, J.; Li, D.; He, C.H.; He, K.; Xue, T.; Wan, L.; Zhang, C.H.; Liu, Q.; Wan, L.; et al. Astrocytic ApoE reprograms neuronal cholesterol metabolism and histone-acetylation-mediated memory. Neuron 2021, 109, 957–970.
- Zou, W.; Zhao, T.; Du, J.; Ji, G.; Li, X.; Ji, S.; Tian, W.; Wang, X.; Hao, A. TIGAR promotes neural stem cell differentiation through acetyl-CoA-mediated histone acetylation. Cell Death Dis. 2019, 10, 198.
- Sun, Y.; Li, T.; Xie, C.; Zhang, Y.; Zhou, K.; Wang, X.; Blomgren, K.; Zhu, C.H. Dichloroacetate treatment improves mitochondrial metabolism and reduces brain injury in neonatal mice. Oncotarget 2016, 7, 31708–31722.
- Luo, C.; Bian, X.; Zhang, Q.; Xia, Z.; Liu, B.; Chn, Q.; Ke, C.; Wu, J.L.; Zhao, Y. Shengui sansheng san ameliorates cerebral energy deficiency via citrate cycle after ischemic stroke. Front. Pharmacol. 2019, 10, 386–392.
- Khoury, N.; Xu, J.; Stegelman, S.D.; Jackson, C.W.; Koronowski, K.B.; Dave, K.R.; Young, J.I.; Perez-Pinzon, M.A. Resveratrol reconditioning induces genomic and metabolic adaptations with long-term window of cerebral ischemic tolerance leading to bioenergetic efficiency. Mol. Neurobiol. 2019, 56, 4549–4565.
- Koronowski, K.B.; Khoury, N.; Saul, N.; Loriz, Z.B.; Cohan, C.H.; Stradecki-Cohan, H.M.; Dave, K.R.; Young, J.I. Neuronal SIRT1 (silent information regulator 2 homologue 1) regulates glycolysis and mediates resveratrol-induced ischemic tolerance. Stroke 2017, 48, 3117–3125.
- Klimaszewska-Łata, J.; Gul-Hinc, S.; Bielarczyk, H.; Ronowska, A.; Zyśk, M.; Grużewska, K.; Pawełczyk, T.; Szutowicz, A. Differential effects of lipopolysaccharide on energy metabolism in murine microglial N9 and cholinergic SN56 neuronal cells. J. Neurochem. 2015, 133, 284–297.
- Gul-Hinc, S.; Michno, A.; Zyśk, M.; Szutowicz, A.; Jankowska-Kulawy, A.; Ronowska, A. Protection of cholinergic neurons against zinc toxicity by glial cells in thiamine-deficient media. Int. J. Mol. Sci. 2021, 22, 13337–13357.
- Ronowska, A.; Gul-Hinc, S.; Bielarczyk, H.; Pawełczyk, T.; Szutowicz, A. Effects of zinc on SN56 cholinergic neuroblastoma cells. J. Neurochem. 2007, 103, 972–983.
- Greco, T.; Vespa, P.M.; Prins, M.L. Alternative substrate metabolism depends on cerebral metabolic state following traumatic brain injury. Exp. Neurol. 2020, 329, 113289.
- Narayanan, S.E.; Rehuman, N.A.; Harilal, S.; Vincent, A.; Rajamma, R.G.; Behl, T.; Uddin, M.S.; Ashraf, G.M.; Mathew, B. Molecular mechanism of zinc neurotoxicity in Alzheimer’s disease. Environ. Sci. Pollut. Res. Int. 2020, 35, 43542–43552.
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791.
- Levenson, C.W. Zinc and traumatic brain injury: From chelation to supplementation. Med. Sci. 2020, 8, 36.
- Ronowska, A.; Gul-Hinc, S.; Michno, A.; Bizon-Zygmańska, D.; Zyśk, M.; Bielarczyk, H.; Szutowicz, A.; Gapys, B.; Jankowska-Kulawy, A. Aggravated effects of coexisting marginal thiamine deficits and zinc excess on SN56 neuronal cells. Nutr. Neurosci. 2021, 6, 432–442.
- Annoni, F.; Peluso, L.; Bogossian, E.G.; Creteur, J.; Zanier, E.R.; Taccone, F.S. Brain protection after anoxic brain injury: Is lactate supplementation helpful ? Cells 2021, 10, 1714.
- Duhaut, D.E.; Heurteaux, C.; Gandin, C.; Ichai, C.; Quintard, H. The antiedematous effect of exogenous lactate therapy in traumatic brain injury: A physiological and mechanistic approach. Neurocrit. Care 2021, 35, 747–755.
- Wang, P.; Chen, M.; Yang, Z.; Yu, T.; Zhu, J.; Zhou, L.; Lin, J.; Fang, X.; Huang, Z.; Jiang, L.; et al. Activation of pyruvate dehydrogenase activity by dichloroacetate improves survival and neurologic outcomes after cardiac arrest in rats. Shock 2018, 49, 704–711.
- Szutowicz, A.; Bielarczyk, H.; Skulimowska, H. Effect of dichloroacetate on acetyl-CoA content and acetylcholine synthesis in rat brain synaptosomes. Neurochem. Res. 1994, 19, 1107–1112.
- Glancy, B.; Balaban, R.S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012, 51, 2959–2973.
- Piao, L.; Fang, Y.H.; Kubler, M.M.; Donnino, M.W.; Sharp, W.W. Enhanced pyruvate dehydrogenase activity improves cardiac outcomes in a murine model of cardiac arrest. PLoS ONE 2017, 12, e0185046.
- Ikeda, K.; Liu, X.; Kida, K.; Marutani, E.; Hirai, S.; Sakaguchi, M.; Andersen, L.W.; Bagchi, A.; Cocchi, M.N.; Berg, K.M.; et al. Thiamine as a neuroprotective agent after cardiac arrest. Resuscitation 2016, 105, 138–144.
- Kho, A.R.; Choi, B.Y.; Lee, S.H.; Hong, D.K.; Jeong, J.H.; Kang, B.S.; Kang, D.H.; Park, K.H.; Park, J.B.; Suh, S.W. The effects of sodium dichloroacetate on mitochondrial dysfunction and neuronal death following hypoglycemia-induced injury. Cells 2019, 8, 405.
- Jakkamsetti, V.; Marin-Valencia, I.; Ma, Q.; Good, L.B.; Terrill, T.; Rajasekaran, K.; Pichumani, K.; Khemtong, C.H.; Hooshyar, M.A.; Sundarrajan, C.H.; et al. Brain metabolism modulates neuronal excitability in a mouse model of pyruvate dehydrogenase deficiency. Sci. Transl. Med. 2019, 11, eaan0457.
- Jakkamsetti, V.; Ma, Q.; Pascual, J.M. A subset of synaptic transmission events is coupled to acetyl coenzyme A production. J. Neurophysiol. 2022, 127, 623–636.
- Chevalier, A.C.; Rosenberger, T.A. Increasing acetyl-CoA metabolism attenuates injury and alters spinal cord lipid content in mice subjected to experimental autoimmune encephalomyelitis. J. Neurochem. 2017, 141, 721–737.
- Della-Flora Nunes, G.; Mueller, L.; Silvestri, N.; Patel, M.S.; Wrabetz, L.; Feltri, M.L.; Poitelon, Y. Acetyl-CoA production from pyruvate is not necessary for preservation of myelin. Glia 2017, 65, 1626–1639.
- Lazzarino, G.; Amorini, A.M.; Signoretti, S.; Musumeci, G.; Lazzarino, G.; Caruso, G.; Pastore, F.S. Pyruvate dehydrogenase and tricarboxylic acid cycle enzymes are sensitive targets of traumatic brain injury induced metabolic derangement. Int. J. Mol. Sci. 2019, 20, 5774.
- Bubber, P.; Haroutunian, V.; Blass, J.P.; Gibson, G.E. Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann. Neurol. 2005, 57, 695–703.
- Huang, Z.; Yan, Q.; Wang, Y.; Zou, Q.; Li, J.; Liu, Z.; Cai, Z. Role of mitochondrial dysfunction in the pathology of amyloid-β. J. Alzheimers Dis. 2020, 78, 505–514.
- Hoshi, M.; Takashima, A.; Murayama, M.; Yoshida, N.; Hohino, T. Nontoxic amyloid beta peptide 1–42 suppresses acetylcholine synthesis. Possible role in cholinergic dysfunction in Alzheimer’s disease. J. Biol. Chem. 1997, 272, 2038–2041.
- Bielarczyk, H.; Jankowska-Kulawy, A.; Höfling, C.; Ronowska, A.; Gul-Hinc, S.; Roβner, S.; Schliebs, R.; Pawełczyk, T.; Szutowicz, A. AβPP-transgenic 2576 mice mimic cell type-specific aspects of acetyl-CoA-linked metabolic deficits in Alzheimer’s disease. J. Alzheimer Dis. 2015, 48, 1083–1094.
- Gandbhir, O.; Sundaram, P. Effect of AmyTrap, an amyloid-β binding drug, on Aβ induced mitochondrial dysfunction and tau phosphorylation in cultured neuroblastoma cells. Metab. Brain Dis. 2020, 35, 923–931.
- Li, S.; Sheng, Z.H. Energy matters: Presynaptic metabolism and the maintenance of synaptic transmission. Nat. Rev. Neurosci. 2022, 23, 4–22.
- Shea, P.A.; Aprison, M.H. The distribution of acetyl-CoA in specific areas of the CNS of the rat as measured by a modification of a radio-enzymatic assay for acetylcholine and choline. J. Neurochem. 1977, 28, 51–58.
- Szutowicz, A.; Tomaszewicz, M.; Jankowska, A.; Kisielevski, J. Acetylocholine synthesis in nerve terminals of diabetic rats. Neuroreport 1994, 5, 2421–2424.
- Szutowicz, A.; Madziar, B.; Pawełczyk, T.; Tomaszewicz, M.; Bielarczyk, H. Effects of NGF on acetylcholine, acetyl-CoA metabolism and viability of differentiated and non-differentiated cholinergic neuroblastoma cells. J. Neurochem. 2004, 90, 952–961.
- Bielarczyk, H.; Jankowska-Kulawy, A.; Gul, S.; Pawełczyk, T.; Szutowicz, A. Phenotype dependent differential effects of interleukin-1β and amyloid-β on viability and cholinergic phenotype of T17 neuroblastoma cells. Neurochem. Int. 2005, 47, 466–473.
- Szutowicz, A.; Bielarczyk, H.; Gul, S.; Zieliński, P.; Pawełczyk, T.; Tomaszewicz, M. Nerve growth factor and acetyl-L-carnitine evoked shifts in acetyl-CoA and cholinergic SN56 cell vulnerability to neurotoxic inputs. J. Neurosci. Res. 2005, 79, 185–192.
- Szutowicz, A.; Bielarczyk, H.; Gul, S.; Ronowska, A.; Pawełczyk, T.; Jankowska-Kulawy, A. Phenotype-dependent susceptibility of cholinergic neuroblastoma cells to neurotoxic inputs. Met. Brain Dis. 2006, 21, 149–161.
- Ronowska, A.; Dyś, A.; Jankowska-Kulawy, A.; Klimaszewska-Łata, J.; Bielarczyk, H.; Romianowski, P.; Pawełczyk, T.; Szutowicz, A. Short-term effects of zinc on acetylcholine metabolism and viability of SN56 cholinergic neuroblastoma cells. Neurochem. Int. 2010, 56, 143–151.
- Jankowska-Kulawy, A.; Bielarczyk, H.; Pawełczyk, T.; Wróblewska, M.; Szutowicz, A. Acetyl-CoA and acetylcholine metabolism in nerve terminal compartment of thiamine deficient rat brain. J. Neurochem. 2010, 115, 333–342.
- Jankowska-Kulawy, A.; Bielarczyk, H.; Pawełczyk, T.; Wróblewska, M.; Szutowicz, A. Acetyl-CoA deficit in brain mitochondria in experimental thiamine deficiency encephalopathy. Neurochem. Int. 2010, 57, 851–856.
- Bizon-Zygmańska, D.; Jankowska-Kulawy, A.; Bielarczyk, H.; Pawełczyk, T.; Ronowska, A.; Marszałł, M.; Szutowicz, A. Acetyl-CoA metabolism in amprolium-evoked thiamine pyrophosphate deficits in cholinergic SN56 neuroblastoma cells. Neurochem. Int. 2011, 59, 208–216.
- Peng, Y.; Li, M.; Clarkson, B.D.; Pehar, M.; Lao, P.J.; Hillmer, A.T.; Barnhart, T.E.; Christian, B.T.; Mitchell, H.A.; Bendlin, B.B.; et al. Deficient import of acetyl-CoA into the ER lumen causes neurodegeneration and propensity to infections, inflammation, and cancer. J. Neurosci. 2014, 34, 6772–6789.
- Hullinger, R.; Li, M.; Wang, J.; Peng, Y.; Dowell, J.A.; Bomba-Warczak, E.; Mitchell, H.A.; Burger, C.; Chapman, E.R.; Denu, J.M.; et al. Increased expression of AT-1/SL33A1 causes an autistic-like phenotype in mice by affecting dendritic branching and spine formation. J. Exp. Med. 2016, 213, 1267–1284.
- Szutowicz, A.; Stępień, M.; Łysiak, W.; Angielski, S. Effect of (-) hydroxycitrate on the activities of ATP citrate lyase and the enzymes of acetyl-CoA metabolism in rat brain. Acta Biochim. Pol. 1976, 23, 227–234.
- Zyśk, M.; Gapys, B.; Ronowska, A.; Gul-Hinc, S.; Erlandsson, A.; Iwanicki, A.; Sakowicz-Burkiewicz, M.; Szutowicz, A.; Bielarczyk, H. Protective effects of voltage-gated calcium channel antagonist against zinc toxicity in SN56 neuroblastoma cholinergic cells. PLoS ONE 2018, 13, e0209363.
- Currais, A.; Huang, L.; Goldberg, J.; Petrascheck, M.; Ates, G.; Pinto-Duarte, A.; Shokhirev, M.N.; Schubert, D.; Maher, P. Elevating acetyl-CoA levels reduces aspects of brain aging. Elife 2019, 8, e47866.
- Dong, Y.; Brewer, G.J. Global metabolic shifts in age and Alzheimer’s disease mouse brains pivot at NAD+/NADH redox sites. J. Alzheimers Dis. 2019, 71, 119–140.
- Ma, Y.; Chen, L.; He, X.X.; Wang, Y.J.; Yu, H.L.; He, Z.X.; Zhang, L.Q.; Zheng, Y.W.; Zhu, X.J. Functional prediction and characterization of Dip2gene in mice. Cell Biol. Int. 2019, 43, 421–428.
- Zyśk, M.; Sakowicz-Burkiewicz, M.; Pikul, P.; Kowalski, R.; Michno, A.; Pawełczyk, T. The impact of acetyl-CoA and aspartate shortages on the N-acetylaspartate level in different models of cholinergic neurons. Antioxidants 2020, 9, 522–545.
- Houston, R.; Sekine, S.; Calderon, M.J.; Seifuddin, F.; Wang, G.; Kawagishi, H.; Malide, D.A.; Li, Y.; Gucek, M.; Pirooznia, M.; et al. Acetylation-mediated remodeling of the nucleolus regulates cellular acetyl-CoA responses. PLoS Biol. 2020, 18, e3000981.
- Lee, J.Y.; Han, S.H.; Park, M.H.; Song, I.S.; Choi, M.K.; Yu, E.; Park, C.M.; Kim, H.J.; Kim, S.H.; Schuchman, E.H.; et al. N-AS-triggered SPMs are direct regulators of microglia in a model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2358.
- Mast, N.; Petrov, A.M.; Prendergast, E.; Bederman, I.; Pikuleva, I.A. Brain acetyl-CoA production and phosphorylation of cytoskeletal proteins are targets of CYP46A1 activity modulation and altered sterol flux. Neurotherapeutics 2021, 18, 2040–2206.
- Huang, W.; Hu, W.; Cai, L.; Zeng, G.; Fang, W.; Dai, X.; Ye, Q.; Chen, X.; Zhang, J. Acetate supplementation produces antidepressant-like effect via enhanced histone acetylation. J. Affect. Disord. 2021, 281, 51–60.
- Suissa, L.; Kotchetkov, P.; Guigonis, J.M.; Doche, E.; Osman, O.; Pourcher, T.; Lindenthal, S. Ingested ketone ester leads to a rapid rise of acetyl-CoA and competes with glucose metabolism in the brain of non-fasted mice. Int. J. Mol. Sci. 2021, 22, 524.
- Wong, V.S.C.; Picci, C.; Swift, M.; Levinson, M.; Willis, D.; Langley, B. α-tubulin acetyltransferase is a novel target mediating neurite growth inhibitory effects of chondroitin sulfate proteoglycans and myelin-associated glycoprotein. eNeuro 2018, 5, e0240-17.
- Hyeonwi-Son, H.; Baek, J.H.; Kang, J.S.; Jung, S.; Chung, H.J.; Kim, H.J. Acutely increased b-hydroxybutyrate plays a role in the prefrontal cortex to escape stressful conditions during the acute stress response. Biochem. Biophys. Res. Commun. 2021, 554, 19–24.
- Kagawa, Y.; Umaru, B.A.; Shima, H.; Ito, R.; Zama, R.; Islam, A.; Kanno, S.I.; Yasui, A.; Sato, S.; Jozaki, K.; et al. FABP7 regulates acetyl-CoA metabolism through the interaction with ACLY in the nucleus of astrocytes. Mol. Neurobiol. 2020, 57, 4891–4910.
- Kagawa, Y.; Umaru, B.A.; Kanamori, M.; Zama, R.; Shil, S.K.; Miyzaki, H.; Kobayashi, S.; Wannakul, T.; Yang, S.; Tominaga, T.; et al. Nuclear FABP7 regulates cell proliferation of wild-type IDH1 glioma through caveolae formation. Mol. Oncol. 2022, 16, 289–306.
- Rigby, M.J.; Orefice, N.S.; Lawton, A.J.; Ma, M.; Shapiro, S.L.; Yi, S.Y.; Dieterich, I.A.; Frelka, A.; Miles, H.N.; Pearce, R.A.; et al. Increased expression of SLC25A1/CIC causes an autistic-like phenotype with altered neuron morphology. Brain 2022, 145, 500–516.