2. Pyroptosis and Inflammation
As mentioned above, the caspase-1/-4/-5/-11-mediated pyroptosis pathways have been involved in the release of proinflammatory cytokines through GSDMD-formed pores
[24][25][26][78,79,80].
Recent research has uncovered that GSDMD can also promote inflammation through various other mechanisms. On the one hand, GSDMD induces the release of IL-1β in a nonpyroptotic manner to exert pyroptosis-like effects. For example, in neutrophils, after the activation and cleavage of GSDMD, GSDMD-N did not migrate to the membrane to form pores, but moved to azure-phagocytic particles and autophagosomes, releasing IL-1β through the formation of pores in the membrane by autophagy
[27][81]. Activation of caspase-8 in intestinal epithelial cells (IECs) can also activate GSDMD, causing the release of IL-1β-containing vesicles through exocytosis. These inflammatory mediators act as exogenous risk factors to further secrete more IL-1β
[28][82]. On the other hand, besides inhibiting GSDMD-N, GSDMD-C can also combine with the p10 fragment produced by caspase-1/-4 self-processing and promote GSDMD cleavage to further enhance pyroptosis
[29][30][83,84]. Additionally, pyroptosis, as one type of programmed death, is closely related to other types of cell death. Multiple studies have demonstrated that caspase-3/-8 can be activated in mice with caspase-1 inactivation or deletion, thereby leading to cytolytic death, which may be caused by the inhibition of GSDMD-related pyroptosis
[31][85]. However, the way of cell death downstream is still controversial. Lee et al. suggested that lytic death is cell necrosis secondary to apoptosis
[32][86], whereas Schneider et al. considered that this may be an inflammatory death that is not identical to either apoptosis or pyroptosis
[33][87].
In the late stages of pyroptosis, the cells swell and membranes rupture, releasing large amounts of inflammatory components including mature IL-1β and IL-18. Although these cytokines were previously assumed to be passively released as a result of cell disintegration, there is now accumulating evidence that secretion precedes plasma membrane rupture in pyroptotic cells. IL-1β binds to the IL-1 receptor to enhance the inflammatory response by triggering NF-κB with accelerated synthesis of proinflammatory agents such as cyclooxygenase-2 and IFN-γ
[34][88]. IL-18 activates the p38-MAPK signaling pathway to increase the release of other inflammatory cytokines, including IL-1α, IL-6, and IL-8
[34][88], which promotes inflammation. Significantly, IL-1β and IL-18 also induce NETosis in neighboring neutrophils, expanding the inflammatory and immune response
[35][36][89,90]. Furthermore, HMBG1 released by pyroptotic cells, on the one hand, triggers DAMPs to promote inflammatory cytokine production, on the other hand, binding to RAGE causes macrophage pyroptosis
[37][38][91,92]. Additionally, pyroptotic cells also release large amounts of ATP, which activates the NLRP3 inflammasome, causing proinflammatory cytokines release
[39][40][93,94].
Collectively, pyroptosis induces cell disintegration and the release of inflammatory cytokines via different mechanisms. Balanced inflammatory responses could activate immune cells and enhance immunity, whereas a sustained activation of pyroptosis-related pathways may promote diseases.
3. Pyroptosis and Inflammasome-Related Disorders
By altering the immune response, activated inflammasomes play essential roles in the context of several inflammatory diseases. Human inflammatory diseases have traditionally been named based on pathologic adaptive immune responses involving excessive antibody responses to self and nonpathogenic external antigens. However, numerous inflammatory diseases fail to merge into this classification, including common diseases associated with obvious tissue inflammation, such as Inflammatory bowel disease (IBD), gout, and Systemic lupus erythematosus (SLE), and also a number of rare genetic disorders associated with systemic and tissue inflammation, namely hereditary febrile diseases. Because experimental and clinical data clearly suggest increased inflammasome activity, several disorders are collectively referred to as inflammasome-related disorders (IRDs), including NLRP3 inflammasome disease, NLRC4 inflammasome disease, pyrin inflammasome disease, and multifactorial inflammasome diseases (
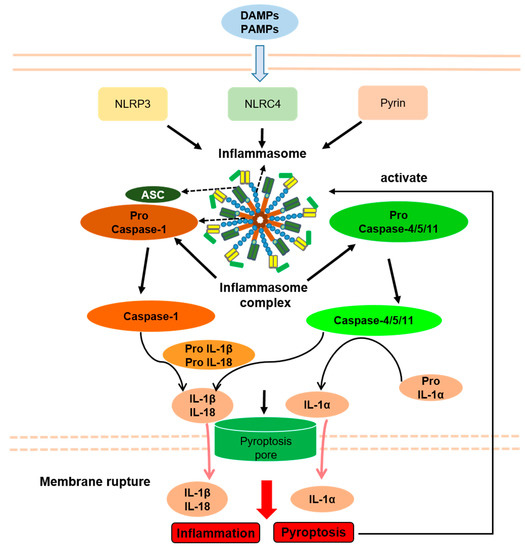
Figure 13).
Figure 13. Inflammasomes and Inflammasome-related disorders association with pyroptosis. PAMPs, pathogen-associated molecular patterns; DAMPs, danger-associated molecular patterns; NLRP3, NLR pyrin domain containing 3; NLRC4, NLR containing a caspase recruitment domain 4; ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain; IL-1β, interleukin-1β; IL-18, interleukin-18; IL-1α, interleukin-1α. Arrows indicate activation or facilitation.
Seemingly, a very common pathogenic mechanism of inflammasomal diseases is a decreased threshold or abnormal continuous activation of inflammasomes caused by gene mutation. Therefore, pyroptosis shares an upstream signaling pathway with IRDs, which may play important roles in the pathogenesis of IRDs. Accordingly, in some IRD animal models, inflammation is alleviated by suppression of pyroptosis by genetic GSDMD deficiency
[41][42][95,96]. Moreover, high levels of IL-1β and IL-18 in IRDs patients may be released through the pores in the membrane formed by pyroptosis and rupture of the membrane, although IL-1β maturation in GSDMD-deficient mice is unaffected
[43][44][49,97]. Importantly, inhibition of IL-1β and IL-18 in inflammasome disease does not completely alleviate inflammation, suggesting that cell death can cause inflammation, which is related to caspase-1 and is likely to be pyroptosis
[45][98].
At present, the relationship between pyroptosis and IRDs has not been fully confirmed as the mechanisms of activation of inflammasomes and subsequent pyroptosis are seemingly different in various etiologies. For example, pyroptosis is induced by bacterial infection, which causes the activation of NLRP3 inflammasomes through a two-step reaction of initiation and activation. However, in IRDs, pyroptosis is triggered by the activation of inflammasomes caused by gene mutation, which usually requires only one step of initiation
[46][99]. Additionally, the activation of the pyrin inflammasomes by bacterial infection requires complete microtubule structures, while the activation of pyrin inflammasome by Mediterranean fever (MEFV) gene mutation does not require microtubule structures
[47][48][100,101]. Furthermore, IL-1β can be released by GSDMD in a manner unrelated to pyroptosis, which is similar to pyroptosis
[28][82]. Therefore, by studying the role of pyroptosis by knocking out GSDMD in IRDs animal models, it may be impossible to determine whether the experimental results are caused by the inhibition of pyroptosis or the inhibition of the release of IL-1β unrelated to pyroptosis. Thus, the specific relationship between IRDs and pyroptosis still needs further study.
3.1. NLRP3 Inflammasome Disease
The NLRP3 inflammasome can be activated either in a canonical or noncanonical manner. On the one hand, it can be activated via caspase-1 by infectious and endogenous ligands such as pore-forming toxins, ATP, and uric acid crystals
[49][50][51][52][102,103,104,105], while on the other hand, caspase-4/-5/-11-mediated LPS sensing also triggers the inflammatory reaction
[53][54][55][56][51,106,107,108]. IRDs related to the NLRP3 inflammasome include both monogenic diseases, such as cryopyrin-associated periodic syndrome (CAPS), and polygenic diseases, such as Crohn’s disease (CD) and gout. This part mainly introduces the relationship between CAPS and pyroptosis
(Table 1).
CAPS is triggered by the continuous activation of NLRP3 inflammasomes due to functionally acquired mutations of the NLRP3 gene, causing persistent caspase-1 activation and disproportionate production of IL-1β and IL-18
[57][58][109,110]. According to the different symptoms, CAPS can be classified into three types: neonatal-onset multisystem inflammatory disease (NOMID), Muckle–Wells syndrome (MWS), and familial cold autoinflammatory syndrome (FCAS). The common clinical symptoms of these diseases include fever, urticaria, and central nervous system inflammation
[59][60][111,112].
Unlike MWS, IL-1 antagonists are only partially effective in NOMID and FCAS patients, suggesting that factors other than IL-1β are involved in the pathogenesis of CAPS
[45][98]. Brydges et al. found that nonapoptotic cell death related to caspase-1 could cause inflammation in the FCAS mouse model, which was speculated to be pyroptosis
[45][98]. Furthermore, studies have shown that protein kinase A (PKA) directly phosphorylates a specific site of NLRP3 to inactivate it and inhibit pyroptosis, and some patients with NOMID have abnormal NLRP3 activation and pyroptosis due to mutations at this site
[61][113]. In an in vivo experiment in NOMID using a mouse model on a GSDMD-deficient background, the results showed that symptoms including skin lesions, splenomegaly, and growth restriction were alleviated, and neutrophil infiltrations in the liver, subcutaneous tissue, and spleen were reduced. It was further confirmed that GSDMD-mediated cell pyroptosis played an important role in the pathogenesis of NOMID, and GSDMD was expected to be a new target for NOMID treatment
[42][96].
3.2. NLRC4 Inflammasome Disease
NLRC4-related inflammasome diseases mainly include autoinflammation with infantile enterocolitis (AIFEC), NOMID, and FCAS4, while only AIFEC has been reported to be associated with pyroptosis
[62][63][114,115]. AIFEC, a newly discovered IRD in 2014, is caused by abnormal activation of the NLRC4 inflammasome due to a functionally acquired mutation in the helical domain 1 domain region of the NLRC4 gene. AIFEC mainly manifests as periodic fever, secretory diarrhea, neonatal colitis, and macrophage activation syndrome. Notably, increased pyroptosis can be detected in peripheral blood of AIFEC patients
[64][65][116,117]. In 2018, Moghaddas et al. found that mutations in the leucine enrichment domain (LRR) of the NLRC4 gene can also cause symptoms similar to AIFEC, but the process of cell pyroptosis caused by LRR mutations does not depend on the involvement of apoptosis proteins (ASC) with lower cytokine response. That means pyroptosis induced by mutations at this site may be downstream of NLRC4 inflammasome and caspase-1 independently of ASC
[66] (Table 1) [118].
3.3. Pyrin Inflammasome Disease
Pyrin-associated autoinflammatory diseases (PAADs) are a group of IRDs caused by the over-activation of the pyrin inflammasome by MEFV gene mutation, which leads to a series of symptoms, such as familial Mediterranean fever (FMF), pyrin-associated autoinflammation with neutrophilic dermatosis (PAAND), chronic aseptic osteomyelitis, and ulcerative dermatitis. Currently, both FMF and PAAND have been reported to be associated with pyroptosis
[67] (Table 1) [119].
FMF encompasses a group of autosomal recessive disorders, and the main clinical manifestations include periodic fever, rash, serositis, and arthritis
[68][120]. The primary cause of FMF is the inactivation of Rho GTPases caused by MEFV gene mutations, which further cause the reduction of the activation threshold of pyrin inflammasomes
[48][101]. Evidence exists that the number of MEFV allele mutations was positively correlated with severity of FMF symptoms and pyroptosis in peripheral blood. Moreover, inhibition of PKN1 and PKN2 proteins (serine/threonine-protein kinase N1/2), which are necessary for pyrin inflammasome activation in peripheral blood of FMF patients profoundly reduced pyroptosis
[48][101]. In recent years, Kanneganti et al. demonstrated that the infection of macrophages in the FMF model with bacteria of the genus Clostridium could cause pyroptosis, accompanied by increased IL-1β secretion. Further in vivo experiments showed that IL-1β levels decreased significantly after GSDMD gene knockout, inflammation was alleviated, and organ-specific inflammatory injuries, such as hepatitis, glomerulonephritis, and colitis were also alleviated
[69][121]. These studies suggest that GSDMD-mediated pyroptosis may play an important role in the pathogenesis of FMF, and GSDMD is expected to be a new target for the treatment of FMF.
PAAND is an autosomal dominant disease with the main clinical manifestations in childhood, which include recurrent neutrophilic dermatitis, periodic fever, joint pain, myalgia, or myositis
[70][122]. Unlike FMF, the pathogenesis of PAAND is caused by mutations in exon 2 of the MEFV gene leading to the disruption of the normal inhibitory state of the pyrin inflammasome and its continued activation, resulting in the massive release of IL-1β and IL-18 and GSDMD-mediated pyroptosis
[71][123]. In patients with PAAND, an increase in pyroptosis can be detected in peripheral blood mononuclear cells
[47][70][72][73][100,122,124,125]. Pyroptosis-mediated membrane pore formation and intracellular DAMP release can cause the production and aggregation of a large number of cytokines in skin and other tissues, which will ultimately lead to neutrophilic dermatitis and inflammation in PAAND patients
[73][125].
3.4. Multifactorial IRDs
The pathogenesis of multifactorial IRDs is seemingly the result of a combination of many elements, such as dietary, environmental, genetic, and immune factors. Thereby, studies indicate that pyroptosis downstream of inflammasome activation may also be related to the pathogenesis of CD, gout and SLE and thus may represent a potential therapeutic target in these rather common diseases
(Table 1).
CD, with abdominal pain, diarrhea, and other gastrointestinal symptoms as the main clinical manifestations, is an inflammatory bowel disease (IBD) with a complex etiology that could be attributed to a combination of genetic and environmental variables. In some patients with CD, the disease is related to alterations in the NOD2 gene
[74][126]. Previous studies have found that the expression of p20 fragments of caspase-1, NLRP3, and GSDMD in IEC and macrophages are increased in patients with CD, suggesting that pyroptosis of both cell types may be closely related to the loss of intestinal mucosal barrier function
[75][76][127,128]. Although the epithelium can eliminate some of the pathogen-infected cells by pyroptosis, this process can support barrier disruption and subsequent inflammation
[77][129]. A multicenter study with a cohort of 100 patients with CD found that the severity of pyroptosis in small intestinal IEC can serve as a potential biomarker for disease severity and predict the therapeutic efficacy of the integrin antagonist vedolizumab
[78][130]. NIMA-related kinase 7 (NEK7) is a necessary enzyme for the activation of the NLRP3 inflammasome
[79][131]. Chen et al. found that activation of NLRP3 in IEC can interact with NEK7 to promote the occurrence of pyroptosis in IEC, which plays a crucial role in the pathogenesis of CD. Knockdown of the NEK7 gene in mice with colitis reduced the expression of pyroptosis-related proteins and intestinal inflammatory symptoms, as well as systemic inflammation
[75][127]. Pyroptosis also plays a proinflammatory role in macrophages associated with CD pathogenesis. A recent study showed that macrophages release nucleoprotein spliceosome-associated protein 130 (SAP130) after intestinal mucosal damage in patients with CD and activate NLRP3/caspase-1, thereby promoting GSDMD cleavage and pyroptosis
[76][128]. However, other studies have shown that although there is less intracellular GSDMD in macrophages compared with IEC, and the pyroptosis mediated by GSDMD mainly plays a proinflammatory role, GSDMD itself can also play a protective role in intestinal inflammation. In a murine colitis model, GSDMD alleviates intestinal inflammatory symptoms by inhibiting the cyclic guanosine phospho-adenosine synthase-interferon gene stimulating factor (cGAS-STING) signaling pathway. Notably, this process was not related to the intestinal flora, and was mainly caused by the excretion of large amounts of potassium ions through pore formation in the membrane
[80][132]. In conclusion, the intracellular GSDMD of macrophages may have both proinflammatory and anti-inflammatory effects, although it seems to have mainly proinflammation in CD.
Gout is a multifactorial IRD caused by the deposition of monosodium urate (MSU) crystals in joints and the surrounding tissues, resulting in systemic inflammation
[81][133]. Although previous studies have shown that MSU-induced inflammation is closely related to the activation of the NLRP3 inflammasome
[52][82][105,134], the role of pyroptosis in the pathogenesis of gout remains controversial
[83][135]. In vitro experiments showed that MSU crystals rapidly increased the expression of GSDMD in mouse macrophages, providing evidence for the excessive occurrence of pyroptosis in gout patients. However, further in vivo experiments showed that MSU crystals did not significantly change cell death or decrease IL-1β levels in mice with GSDMD, caspase-1, or MLKL deficiency. Moreover, inhibition of NLRP3 inflammasome by extracellular high potassium did not significantly affect the occurrence of cell death, suggesting that MSU-induced cell death is not mediated by pyroptosis or necroptosis
[84][136]. However, Li et al. suggested that pyroptosis exerts an important role in the development of gout. Purine guanosine monophosphate receptor P2Y14R negatively regulates NLRP3 inflammasomes, and in mice with knockout of the P2Y14R gene, NLRP3 expression and MSU-related pyroptosis were downregulated, while gout symptoms were alleviated
[85][137]. In conclusion, the role of pyroptosis in the pathogenesis of gout is still controversial, and whether GSDMD can be a target for the treatment of gout needs further investigation.