Cancer cachexia (CC) is a multifactorial syndrome characterized by a significant reduction in body weight that is predominantly caused by the loss of skeletal muscle and adipose tissue. Although the ill effects of cachexia are well known, the condition has been largely overlooked, in part due to its complex etiology, heterogeneity in mediators, and the involvement of diverse signaling pathways. For a long time, inflammatory factors have been the focus when developing therapeutics for the treatment of CC. Despite promising pre-clinical results, they have not yet advanced to the clinic. Developing new therapies requires a comprehensive understanding of how deregulated signaling leads to catabolic gene expression that underlies muscle wasting.
- cancer
- cytokine signaling
- muscle wasting
- transcription factors
- epigenetics
1. Introduction
2. Mediators of Skeletal Muscle Wasting
In general, most cancer patients suffer from anorexia (loss of appetite). Even sufficient nutritional support fails to reverse the progressive weight loss. The metabolic changes differ between anorexic and cachectic patients [1,5][1][5]. This indicates that reduced food intake alone is less likely to contribute to the complex muscle-wasting process. Parabiosis studies in animal models provide evidence for the humoral mediation of CC [6,7][6][7]. These studies indicate that CC might be driven, at least in part, by circulating factors released by either host or tumor cells. Among these, inflammatory cytokines such as tumor necrosis factor alpha (TNFα), Interleukin-6 (IL-6) and transforming growth factor β (TGFβ) family members have been studied in detail. In CC, altered cytokine levels tilt the balance between muscle anabolic and catabolic genes towards catabolism, resulting in the degradation of muscle proteins. Cytokine-mediated signaling leading to the transcriptional regulation of muscle catabolic genes is critical in cachexia. In particular, the expression of the E3 ubiquitin ligase muscle RING finger containing protein 1 (MURF1) and muscle atrophy F box protein (MAFbx, also known as Atrogin-1) are central to ubiquitin-mediated myofibrillar protein degradation in cachectic skeletal muscle [8]. Several signaling pathways converge to activate the MURF1 and Atrogin-1 mediated degradation of skeletal muscle proteins that causes muscle wasting (Figure 1). Atrogin-1 controls the activity of the translation machinery protein eIF3f by ubiquitination [9]. Silencing Atrogin-1 leads to the upregulation of MyoD and the downregulation of myostatin, a negative regulator of muscle mass [10].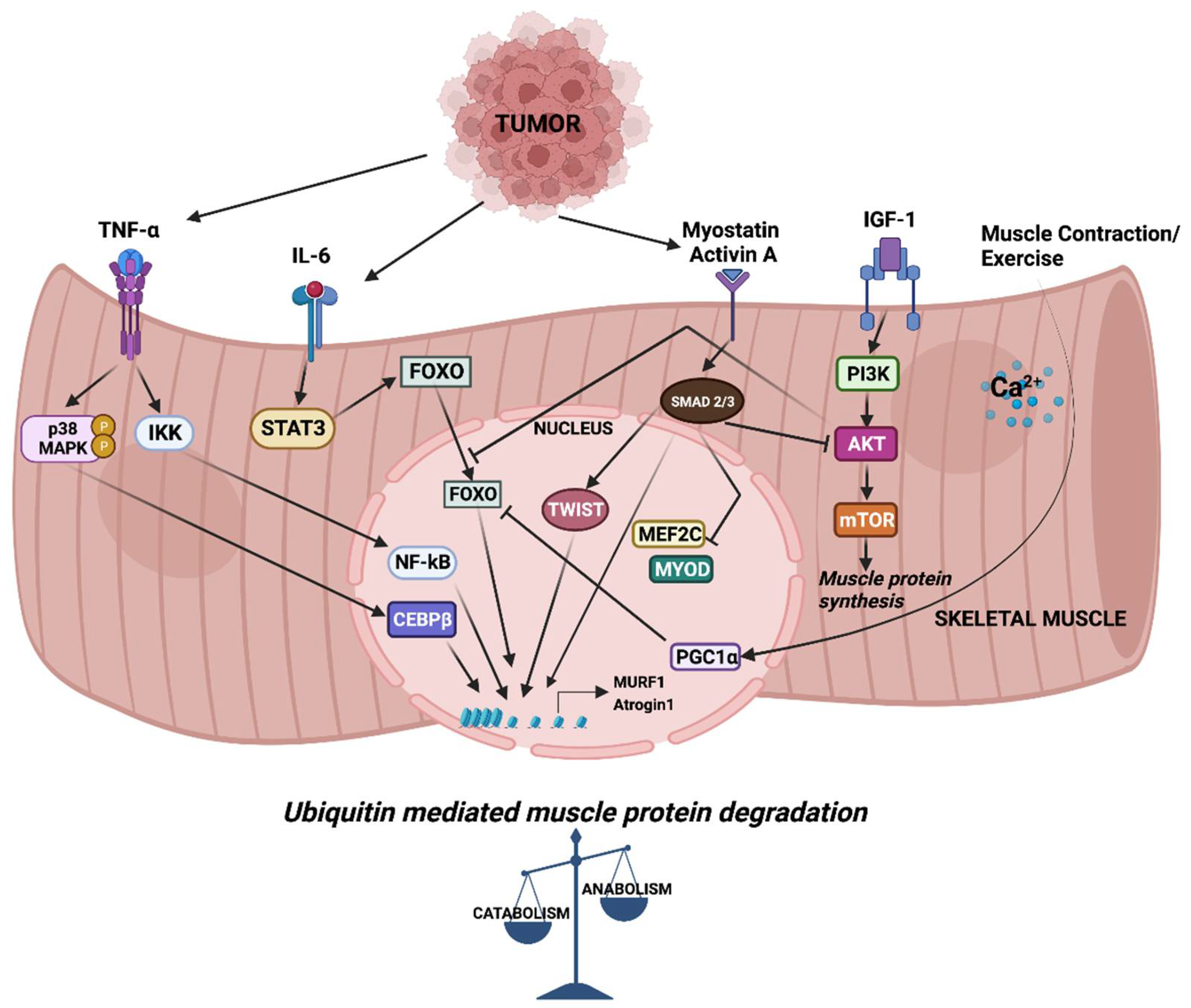
3. Epigenetic Regulation of Muscle Catabolic Genes
The activity of FOXO transcription factors is tightly regulated by epigenetic factors (Figure 32). The acetylation of FOXO by p300-CBP acetyltransferase prevents nuclear localization and reduces its transcriptional activity [112][15]. HDAC1 increases FOXO activity, causing the elevated expression of atrogenes including MURF1 and Atrogin-1, and leads to muscle atrophy [113][16]. Trichostatin A (TSA), a HDAC inhibitor, counteracts unloading-induced muscle wasting. TSA treatment partly prevents the loss of type I and type IIa muscle fiber size in mice. Furthermore, HDAC4 interacts with and deacetylates FOXO3, leading to its activation, and thereby promotes denervation-induced muscle wasting in mice [114][17]. N6-methyladenosine (m6A) is one of the most abundantly studied post-transcriptional modifications of eukaryotic mRNA. Using a denervation-induced muscle atrophy mouse model, the m6A demethylase ALKB homologue 5 (ALKBH5) was shown to stabilize HDAC4 mRNA [115][18].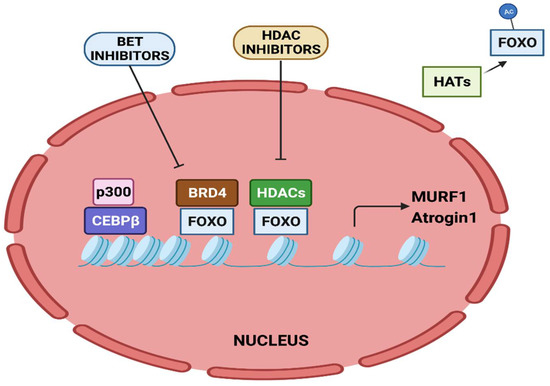
References
- Tisdale, M.J. Cachexia in Cancer Patients. Nat. Rev. Cancer 2002, 2, 862–871.
- Tisdale, M.J. Mechanisms of Cancer Cachexia. Physiol. Rev. 2009, 89, 381–410.
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and Classification of Cancer Cachexia: An International Consensus. Lancet Oncol. 2011, 12, 489–495.
- Fearon, K.C.H. Cancer Cachexia: Developing Multimodal Therapy for a Multidimensional Problem. Eur. J. Cancer 2008, 44, 1124–1132.
- Evans, W.K.; Makuch, R.; Clamon, G.H.; Feld, R.; Weiner, R.S.; Moran, E.; Blum, R.; Shepherd, F.A.; Jeejeebhoy, K.N.; DeWys, W.D. Limited Impact of Total Parenteral Nutrition on Nutritional Status during Treatment for Small Cell Lung Cancer. Cancer Res. 1985, 45, 3347–3353.
- Norton, J.A.; Moley, J.F.; Green, M.V.; Carson, R.E.; Morrison, S.D. Parabiotic Transfer of Cancer Anorexia/Cachexia in Male Rats. Cancer Res. 1985, 45, 5547–5552.
- Tessitore, L.; Costelli, P.; Baccino, F.M. Humoral Mediation for Cachexia in Tumour-Bearing Rats. Br. J. Cancer 1993, 67, 15–23.
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of Ubiquitin Ligases Required for Skeletal Muscle Atrophy. Science 2001, 294, 1704–1708.
- Lagirand-Cantaloube, J.; Offner, N.; Csibi, A.; Leibovitch, M.P.; Batonnet-Pichon, S.; Tintignac, L.A.; Segura, C.T.; Leibovitch, S.A. The Initiation Factor EIF3-f Is a Major Target for Atrogin1/MAFbx Function in Skeletal Muscle Atrophy. EMBO J. 2008, 27, 1266–1276.
- Cong, H.; Sun, L.; Liu, C.; Tien, P. Inhibition of Atrogin-1/MAFbx Expression by Adenovirus-Delivered Small Hairpin RNAs Attenuates Muscle Atrophy in Fasting Mice. Hum. Gene Ther. 2011, 22, 313–324.
- Khal, J.; Hine, A.V.; Fearon, K.C.H.; Dejong, C.H.C.; Tisdale, M.J. Increased Expression of Proteasome Subunits in Skeletal Muscle of Cancer Patients with Weight Loss. Int. J. Biochem. Cell Biol. 2005, 37, 2196–2206.
- Bossola, M.; Muscaritoli, M.; Costelli, P.; Grieco, G.; Bonelli, G.; Pacelli, F.; Rossi Fanelli, F.; Doglietto, G.B.; Baccino, F.M. Increased Muscle Proteasome Activity Correlates with Disease Severity in Gastric Cancer Patients. Ann. Surg. 2003, 237, 384–389.
- Khal, J.; Wyke, S.M.; Russell, S.T.; Hine, A.V.; Tisdale, M.J. Expression of the Ubiquitin-Proteasome Pathway and Muscle Loss in Experimental Cancer Cachexia. Br. J. Cancer 2005, 93, 774–780.
- Zhang, L.; Tang, H.; Kou, Y.; Li, R.; Zheng, Y.; Wang, Q.; Zhou, X.; Jin, L. MG132-Mediated Inhibition of the Ubiquitin-Proteasome Pathway Ameliorates Cancer Cachexia. J. Cancer Res. Clin. Oncol. 2013, 139, 1105–1115.
- Senf, S.M.; Sandesara, P.B.; Reed, S.A.; Judge, A.R. P300 Acetyltransferase Activity Differentially Regulates the Localization and Activity of the FOXO Homologues in Skeletal Muscle. Am. J. Physiol.-Cell Physiol. 2011, 300, C1490–C1501.
- Beharry, A.W.; Sandesara, P.B.; Roberts, B.M.; Ferreira, L.F.; Senf, S.M.; Judge, A.R. HDAC1 Activates FoxO and Is Both Sufficient and Required for Skeletal Muscle Atrophy. J. Cell Sci. 2014, 127, 1441–1453.
- Dupré-Aucouturier, S.; Castells, J.; Freyssenet, D.; Desplanches, D. Trichostatin A, a Histone Deacetylase Inhibitor, Modulates Unloaded-Induced Skeletal Muscle Atrophy. J. Appl. Physiol. (1985) 2015, 119, 342–351.
- Liu, Y.; Zhou, T.; Wang, Q.; Fu, R.; Zhang, Z.; Chen, N.; Li, Z.; Gao, G.; Peng, S.; Yang, D. M6 A Demethylase ALKBH5 Drives Denervation-Induced Muscle Atrophy by Targeting HDAC4 to Activate FoxO3 Signalling. J. Cachexia Sarcopenia Muscle 2022, 13, 1210–1223.
- Segatto, M.; Fittipaldi, R.; Pin, F.; Sartori, R.; Dae Ko, K.; Zare, H.; Fenizia, C.; Zanchettin, G.; Pierobon, E.S.; Hatakeyama, S.; et al. Epigenetic Targeting of Bromodomain Protein BRD4 Counteracts Cancer Cachexia and Prolongs Survival. Nat. Commun. 2017, 8, 1707.
- Parajuli, P.; Kumar, S.; Loumaye, A.; Singh, P.; Eragamreddy, S.; Nguyen, T.L.; Ozkan, S.; Razzaque, M.S.; Prunier, C.; Thissen, J.-P.; et al. Twist1 Activation in Muscle Progenitor Cells Causes Muscle Loss Akin to Cancer Cachexia. Dev. Cell 2018, 45, 712–725.e6.
- Nayak, A.; Lopez-Davila, A.J.; Kefalakes, E.; Holler, T.; Kraft, T.; Amrute-Nayak, M. Regulation of SETD7 Methyltransferase by SENP3 Is Crucial for Sarcomere Organization and Cachexia. Cell Rep. 2019, 27, 2725–2736.e4.
- Amrute-Nayak, M.; Pegoli, G.; Holler, T.; Lopez-Davila, A.J.; Lanzuolo, C.; Nayak, A. Chemotherapy Triggers Cachexia by Deregulating Synergetic Function of Histone-Modifying Enzymes. J. Cachexia Sarcopenia Muscle 2021, 12, 159–176.
- Kottorou, A.; Dimitrakopoulos, F.-I.; Tsezou, A. Non-Coding RNAs in Cancer-Associated Cachexia: Clinical Implications and Future Perspectives. Transl. Oncol. 2021, 14, 101101.
- Narasimhan, A.; Ghosh, S.; Stretch, C.; Greiner, R.; Bathe, O.F.; Baracos, V.; Damaraju, S. Small RNAome Profiling from Human Skeletal Muscle: Novel MiRNAs and Their Targets Associated with Cancer Cachexia. J. Cachexia Sarcopenia Muscle 2017, 8, 405–416.
- Li, X.; Du, L.; Liu, Q.; Lu, Z. MicroRNAs: Novel Players in the Diagnosis and Treatment of Cancer Cachexia (Review). Exp. Ther. Med. 2022, 24, 446.
- Fernandez, G.J.; Ferreira, J.H.; Vechetti, I.J., Jr.; De Moraes, L.N.; Cury, S.S.; Freire, P.P.; Gutiérrez, J.; Ferretti, R.; Dal-Pai-Silva, M.; Carvalho, R.F.; et al. MicroRNA-MRNA Co-Sequencing Identifies Transcriptional and Post-Transcriptional Regulatory Networks Underlying Muscle Wasting in Cancer Cachexia. Front. Genet. 2020, 11, 541.
- Han, J.; Shen, L.; Zhan, Z.; Liu, Y.; Zhang, C.; Guo, R.; Luo, Y.; Xie, Z.; Feng, Y.; Wu, G. The Long Noncoding RNA MALAT1 Modulates Adipose Loss in Cancer-Associated Cachexia by Suppressing Adipogenesis through PPAR-γ. Nutr. Metab. 2021, 18, 27.
- Shi, X.; Yang, J.; Liu, M.; Zhang, Y.; Zhou, Z.; Luo, W.; Fung, K.-M.; Xu, C.; Bronze, M.S.; Houchen, C.W.; et al. Circular RNA ANAPC7 Inhibits Tumor Growth and Muscle Wasting via PHLPP2-AKT-TGF-β Signaling Axis in Pancreatic Cancer. Gastroenterology 2022, 162, 2004–2017.e2.
- He, W.A.; Calore, F.; Londhe, P.; Canella, A.; Guttridge, D.C.; Croce, C.M. Microvesicles Containing MiRNAs Promote Muscle Cell Death in Cancer Cachexia via TLR7. Proc. Natl. Acad. Sci. USA 2014, 111, 4525–4529.
- Chacon-Cabrera, A.; Fermoselle, C.; Salmela, I.; Yelamos, J.; Barreiro, E. MicroRNA Expression and Protein Acetylation Pattern in Respiratory and Limb Muscles of Parp-1(-/-) and Parp-2(-/-) Mice with Lung Cancer Cachexia. Biochim. Biophys. Acta 2015, 1850, 2530–2543.