Alzheimer’s disease (AD) is the most common neurodegenerative disorder and its prevalence is increasing. Very few drugs effectively reduce AD symptoms and thus, a better understanding of its pathophysiology is vital to design new effective schemes. Presymptomatic neuronal damage caused by the accumulation of Amyloid β peptide and Tau protein abnormalities remains a challenge, despite recent efforts in drug development. Importantly, therapeutic targets, biomarkers, and diagnostic techniques have emerged to detect and treat AD. Of note, the compromised blood-brain barrier (BBB) and peripheral inflammation in AD are becoming more evident, being harmful factors that contribute to the development of the disease. Perspectives from different pre-clinical and clinical studies link peripheral inflammation with the onset and progression of AD.
- Alzheimer’s disease
- blood-brain barrier
- blood-borne factors
- neurovascular unit
- exercise
1. Introduction
2. The Role of Aβ in AD
Amyloid precursor protein (APP) is a transmembrane protein with three domains, extracellular, transmembrane, and intracellular [10][6]. Soluble APP (sAPP) is involved in cell survival, neurite growth, synaptogenesis, and synaptic plasticity [11,12][7][8]. APP can be cleaved by several secretases and the non-amyloid processing involves α-and γ-secretases. In amyloidogenic forms, β-secretase (BACE1), rather than α-secretase, is the main protease involved, producing mostly Aβ1-40 and, to a lesser extent, the more amyloidogenic form Aβ1-42. Aβ1-40 tends to accumulate in the vasculature, while Aβ1-42 constitutes the predominant form in amyloid plaques [13][9]. Monomers, oligomers, fibrils, and amyloid plaques are the different conformations of Aβ aggregation in the brains of patients with AD. Aβ does not adopt a single folded form but acquires a set of conformations prone to aggregation in the form of Aβ oligomers, and these are transient forms between monomers and fibrils [14][10]. Oligomers and fibrils are the most toxic arrangements for forming Aβ [15][11]. Importantly, ε4 allele of apolipoprotein E (APOEε4) is the main genetic risk of late-onset AD. Apo-E proteins participate in Aβ clearance and Apo-E4 is less efficient in performing this process than other isoforms such as Apo-E3, suggesting that its participation in Aβ accumulation is in accordance with the increased deposition of Aβ observed in the brain of APOEε4 allele carriers [19][12]. AD pathology investigation has been widely focused on Aβ theory, however, there are still no successful therapies to treat AD by targeting Aβ and recently the theory was challenged since some inconsistencies were found in the research of Aβ*56 isoform [20][13]; therefore, the role of Aβ*56 in AD should be carefully considered and other strategies such as targeting Tau or microglia activity need to be addressed as therapeutic alternatives to Aβ targeting [21][14].3. Tau and AD
Tau neurofibrillary tangles are closely related to neuronal loss and clinical symptoms of AD [16][15]. Although Aβ can initiate a cascade of events related to AD onset, Tau impairment is probably the main effector that induces neurodegeneration. Tau is involved in microtubule stabilization and dynamics, myelination, axonal transport, neurogenesis, neuronal excitability, glucose metabolism, DNA protection, iron homeostasis, motor, learning, and memory functions [22][16]. The accumulation of Aβ is a hallmark of AD, while Tau pathology also exists in other tauopathies [23][17]. Tau is predominantly expressed in the adult human central and peripheral nervous system, with different isoforms in the central nervous system (CNS) produced through alternative splicing of the Microtubule-associated protein tau (MAPT) comprising 16 exons located on chromosome 17q21.3 [24][18]. Tau is most abundant in the axons of nerve cells. In the brain, Tau has several post-translational modifications and the most studied is its phosphorylation which negatively regulates its ability to interact with microtubules. Tau is primarily an intracellular protein, but it is also present in the extracellular space and interstitial fluid [25][19]. Tau is phosphorylated in the AD human brain forming intraneuronal aggregates known as NFT [26][20], although not all phosphorylated Tau is aggregated [27][21]. In AD, Tau aggregates follow a well-defined pattern, which begins in the entorhinal cortex and hippocampus [25][19]. Tau pathology can also be mediated by Aβ oligomerization which induces Tau oligomerization in vitro [28][22]. Moreover, Aβ deposition was associated with increased phosphorylated Tau in the cerebrospinal fluid from AD patients and the 5xFAD mouse model [29][23].4. The Synergy between Aβ and Tau
Aβ depositions and NFT are hallmarks of AD and act through different mechanisms to induce neurotoxicity. Nevertheless, therapies that target Aβ plaques or Tau do not have satisfactory results [30][24]. Interestingly, Tau is a mediator of Aβ plaques cytotoxicity [31][25] and Aβ and Tau may interact via intermediate molecules (e.g., some kinases) [32][26]. An interplay between Aβ and Tau amplifies toxic effects rather than a mode of interaction [33][27]. The interplay between Aβ and Tau has been suggested in AD pathology. Studies carried out with Tau in its isolated form of human samples with or without Aβ plaques showed that Tau aggregation properties were enhanced in Tau derived from subjects containing Aβ plaques. Aβ plaques and Tau synergistic interaction also affects intracellular targets (e.g., mitochondria), thus amplifying neurotoxic effects. Mitochondrial alterations are implicated in AD [42][28] and Aβ plaques, NFT, and neurotoxicity are related with mitochondrial alterations [43][29]. Several proteins interact with Aβ plaques and phosphorylated Tau (e.g., Dynamin-1-like protein Drp1 (DNM1L), caspase-3, HSD17B10), leading to mitochondrial fragmentation and neuronal death [30,44][24][30]. Finally, Aβ and Tau coexist in pathological sites in AD [30,45][24][31].5. Neuroinflammation in Alzheimer’s Disease
In recent years, the sustained immune response in the brain of patients with AD has been considered a substantial part of the central pathology of AD [46[32][33][34][35][36],47,48,49,50], which is observed in post-mortem brains and AD preclinical models [51,52,53,54,55,56][37][38][39][40][41][42]. Sustained activation of microglia in the brain and other immune cells has been observed, which exacerbates Aβ and Tau pathologies and could be a link in the pathogenesis of AD [57][43]. Neuroinflammation is an inflammatory response within the brain and spinal cord [58][44]. Infections, toxins, and other injuries activate acute inflammation in the brain. Chronic neuroinflammation occurs when there is a lack of balance in the anti-inflammatory and proinflammatory response, as with AD, which presents activated microglia and cytokines [59,60][45][46]. Neuroinflammation is not unique to AD and is also seen in Parkinson’s disease (PD) [61[47][48],62], chronic traumatic encephalopathy [63][49], amyotrophic lateral sclerosis [64][50], and multiple sclerosis [65][51]. Equivalently, it is proposed that activation of the innate immune system is essential at the onset and progression of AD. Several genes that code for receptors with an immunological role, such as CD33, CLU, CR1, and TREM2, are altered in AD [70,71,72][52][53][54]. Various genes that regulate the immune system inside and outside the CNS, such as BLNK, GRN, HEXB, PYDC1, SYK, and SLC2A5, are related to this disease [73,74][55][56]. These changes in gene expression suggest a role of the central and peripheral immune systems in AD.6. The BBB Barrier in AD
Three brain barriers protect and maintain the brain microenvironment: the arachnoid barrier, the blood-cerebrospinal fluid barrier, and the BBB [75,76][57][58]. The BBB is an interface between the peripheral circulation and the CNS. It is a semi-permeable structural and chemical barrier, highly selective, that separates the circulating blood from the brain, and the extracellular fluid [77][59]. The neurovascular unit is the fundamental component of the BBB and is composed of capillary endothelial cells (its main structure), astrocytes, pericytes, neurons, and tight junctions [78][60]. In a healthy brain, the BBB does not allow the entrance of several immune cells, hydrophilic molecules, and large proteins into the brain and vice versa [79][61]. The permeability of BBB is not associated with the molecular size itself: glucose and amino acids pass through the BBB more easily than many ions [80][62], and permeability increases in the presence of cerebral edema, brain tumors, ionizing radiation injury, inflammation, and other pathological conditions, allowing certain toxic substances to enter brain tissue and ultimately resulting in damage to the CNS [81][63]. Strokes, immunoneurological diseases, and multiple system atrophy significantly alter the BBB function [82][64]. However, not all substances are harmful; some antibodies reach injured regions of the brain and contribute to its restoration [83][65]. It is noteworthy to highlight that many plasma proteins can reach the healthy brain mainly through Clathrin transport [84][66], thus suggesting the presence of plasma-derived biomolecules with regenerative properties that could be tested to treat ailments such as AD. A large body of evidence indicates that the BBB is impaired in neurodegenerative conditions [85][67] such as AD and PD. AD patients have shown a compromised BBB since leakage of plasma proteins and blood cells have been found in brain regions such as the prefrontal cortex, entorhinal cortex, and hippocampus of post-mortem tissues when compared with control brains [85,86,87,88][67][68][69][70]. Imaging studies further support the disruption of the BBB on AD even at early stages [89][71]; in line with these data, brain endothelial cells derived from induced pluripotent stem cells (iPSCs) of AD patients display aberrant properties such as high permeability and altered expression of tight junctions [90][72]. Additionally, animal studies have provided consistent results since rodent models of AD also display a disrupted BBB [91][73]. Remarkably, rodent AD models suggest an increased vulnerability of BBB to be compromised after an inflammatory insult; therefore, inflammation could contribute to BBB disruption [91][73]. Excessive Aβ generation and its deposition in the brain also contribute to the disruption of BBB [92][74] by increasing the damage to the neurovascular unit [8][75]. Increased BBB permeability in AD could be related to pericyte loss possibly mediated by MMP-9 in ApoE4 carriers. Aβ deposition also is related with pericyte death and the disruption of astrocytes end feet and arterioles coupling. Furthermore, Aβ plaques and NFTs are associated with thickening of cerebral blood vessels leading to hemorrhages [93][76]. Vascular basement membrane thickening in AD is observed in brain regions such as the cerebral cortex, hippocampus, and thalamus mainly in the parenchymal basement membrane suggesting the participation of astrocytes since they are associated with the synthesis of this basement membrane. Furthermore, other studies reported the altered synthesis and degradation of basement membrane components (e.g., collagen IV) [94,95][77][78]. Increased monocyte trafficking through the BBB in response to Aβ has been identified in the peripheral circulation or brain parenchyma and associated with the pathophysiology of Aβ-related vascular disorder [96][79]. In addition, Aβ induces the expression of vascular adhesion molecules, promoting leukocyte adhesion and transmigration during the pathological process of AD [97][80]. In animal models it has been shown that Aβ deposits were able to increase the chemoattraction and permeability of neutrophils and bone marrow-derived microglia as a cellular mechanism for the restriction and elimination AB deposits [98,99][81][82]. Additionally, it was observed that monocytes were attracted only to the luminal wall of veins with Aβ deposits where they engulf Aβ for its elimination. This process occurs mainly in the cortex and hippocampus [100][83]. BBB damage induced by Aβ results not only from direct toxicology in endothelial cells, astrocytes, and pericytes but also from indirect neuroinflammation and oxidative stress triggered by Aβ accumulation, as well as from the impaired interaction between different cellular components in BBB [93][76]. Moreover, Aβ1-42 decreases the expression of tight junction proteins occluding and claudin-5 in endothelial cells and related tight junction proteins such as ZO-1; conversely, Aβ1-42 increases MMP-2 and MMP-9 activity that subsequently enhances endothelial permeability. Interestingly, the inactivation of the receptor for advanced glycation end products (RAGE) attenuates these impairments, indicating that RAGE mediates the endothelial cell alterations derived from the exposure to Aβ1-42 (Figure 1) [101][84].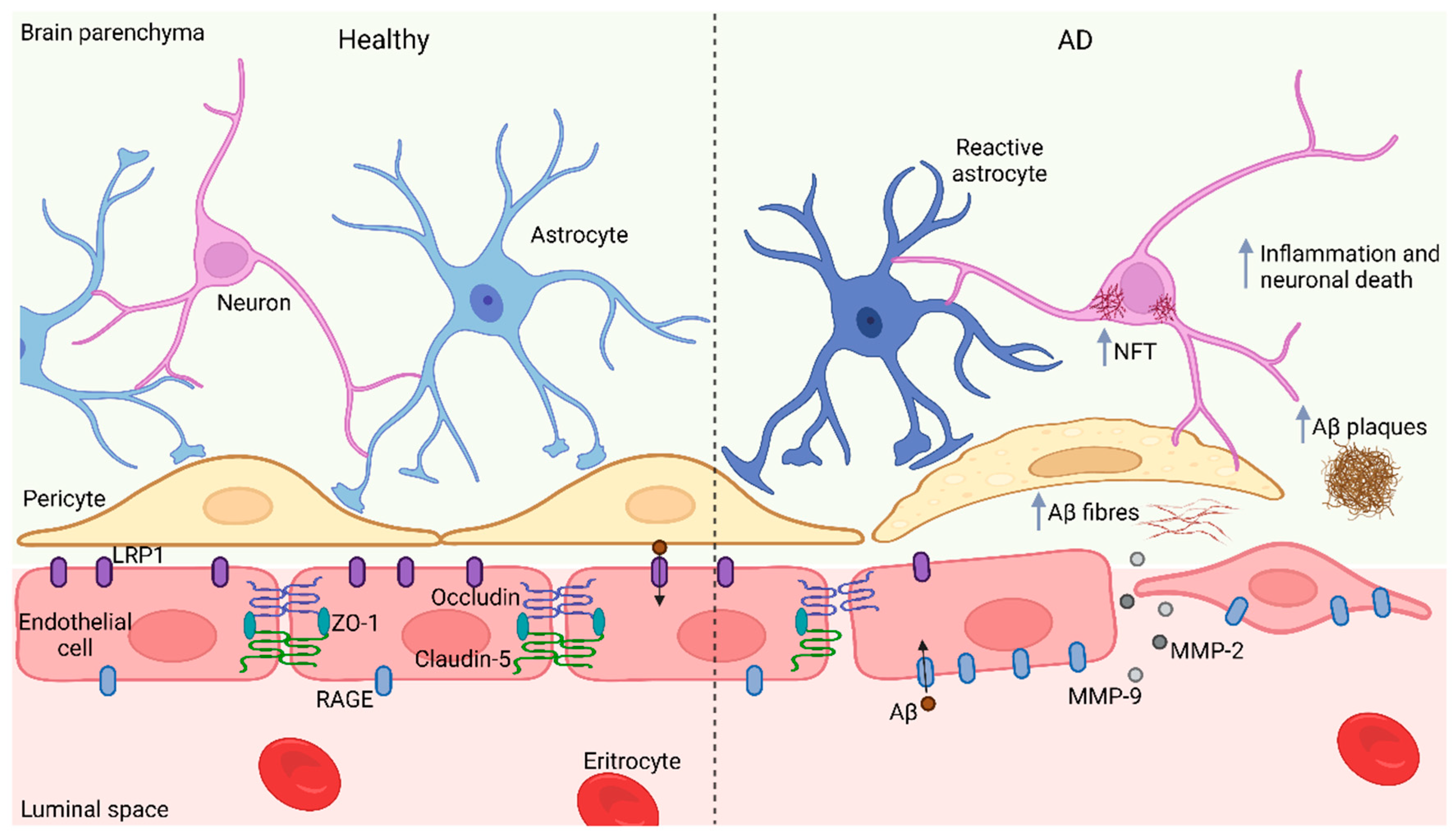
7. BBB Impairment: An Opportunity to Treat AD?
In contrast with the harmful agents of the milieu that could reach the brain in AD, the free entrance of protective factors to the brain parenchyma due to a compromised BBB in AD could also be possible; hence, their capability to cross the BBB should be further evaluated and considered as a therapeutic strategy. Some of these factors could be present in blood from young individuals since pre-clinical studies have reported the recovery and improvement of AD mice after receiving plasma from younger animals [108][85]. Unfortunately, there are still no beneficial effects detected in AD patients after receiving plasma from young volunteers [109][86]. The divergence of these results could be explained by different causes such as the fact that mice AD models do not entirely reproduce the AD, therefore the animal results could differ from humans. On the other hand, it should also be considered that the effect of beneficial factors could be masked by other unspecific molecules present in plasma. Therefore, there is a need to identify and isolate the specific plasma molecules from human origin with the capability to prevent neurodegeneration in AD. The identification of beneficial factors in the blood represents a formidable challenge given the extreme complexity of blood which has hampered the identification of especially neuroprotective and rejuvenating cues. Recent pioneering studies identified the protein Clusterin (CLU), upregulated in plasma from exercised rodents and humans. Remarkably, intravenous administration of CLU induced an anti-inflammatory phenotype in the brain endothelial cells of the hippocampus of transgenic AD mice, suggesting its potential as a therapeutic agent to treat this neurodegenerative condition [110][87]. In line with these findings, another recent work identified the Glycosylphosphatidylinositol specific phospholipase D1 (Gpld1) as a beneficial factor in the blood of exercised subjects that improves cognitive skills and plasticity mechanisms in the brain when transferred to sedentary rodents [111][88]. Interestingly, there is a correlation between exercise routines and the amelioration of symptoms in AD [112,113][89][90]; this information suggests that Gpld1 and CLU should be considered to treat AD since they could be administered intravenously and freely reach the affected areas of the brain. Given this evidence, it is noteworthy to highlight the importance of identifying other blood-borne factors of either young or exercised individuals with neuroprotective properties that could contribute to the improvement of AD treatment. Although promising, several contrasting reports suggest that BBB remains unaltered in AD and that drugs and blood-borne factors cannot cross it [114,115,116][91][92][93]. If this is the case, there is a need to develop therapeutic strategies to overcome this obstacle. Several biologic molecules, such as antibodies capable of crossing the BBB, have been conjugated with trophic factors and have the potential to treat AD. Detailed and recent information regarding this exciting field can be found elsewhere [117][94]. However, it is also possible that some neuroprotective factors do not cross the BBB; instead, they could interact and stimulate membrane receptors on brain endothelial cells from the luminal space and initiate beneficial effects by secreting cues to the brain parenchyma. CLU binds to the brain blood vessels when administered intravenously [110][87] therefore, the possibility that this protein does not cross the BBB cannot be excluded.References
- Dementia Statistics|Alzheimer’s Disease International (ADI). Available online: https://www.alzint.org/about/dementia-facts-figures/dementia-statistics/ (accessed on 17 May 2022).
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213.
- Dhana, K.; Franco, O.H.; Ritz, E.M.; Ford, C.N.; Desai, P.; Krueger, K.R.; Holland, T.M.; Dhana, A.; Liu, X.; Aggarwal, N.T.; et al. Healthy Lifestyle and Life Expectancy with and without Alzheimer’s Dementia: Population Based Cohort Study. BMJ 2022, 377, e068390.
- Murphy, M.P.; Levine, H. Alzheimer’s Disease and the Amyloid-β Peptide. J. Alzheimer’s Dis. 2010, 19, 311–323.
- Ksiezak-Reding, H.; Liu, W.K.; Yen, S.H. Phosphate Analysis and Dephosphorylation of Modified Tau Associated with Paired Helical Filaments. Brain Res. 1992, 597, 209–219.
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Müller-Hill, B. The Precursor of Alzheimer’s Disease Amyloid A4 Protein Resembles a Cell-Surface Receptor. Nature 1987, 325, 733–736.
- Müller, U.C.; Deller, T.; Korte, M. Not Just Amyloid: Physiological Functions of the Amyloid Precursor Protein Family. Nat. Rev. Neurosci. 2017, 18, 281–298.
- Rice, H.C.; de Malmazet, D.; Schreurs, A.; Frere, S.; van Molle, I.; Volkov, A.N.; Creemers, E.; Vertkin, I.; Nys, J.; Ranaivoson, F.M.; et al. Secreted Amyloid-β Precursor Protein Functions as a GABA B R1a Ligand to Modulate Synaptic Transmission. Science 2019, 363, 123.
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An Overview of APP Processing Enzymes and Products. Neuromolecular Med. 2010, 12, 1.
- Ball, K.A.; Phillips, A.H.; Nerenberg, P.S.; Fawzi, N.L.; Wemmer, D.E.; Head-Gordon, T. Homogeneous and Heterogeneous Tertiary Structure Ensembles of Amyloid-β Peptides. Biochemistry 2011, 50, 7612–7628.
- Benilova, I.; Karran, E.; de Strooper, B. The Toxic Aβ Oligomer and Alzheimer’s Disease: An Emperor in Need of Clothes. Nat. Neurosci. 2012, 15, 349–357.
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer Disease: Risk, Mechanisms and Therapy. Nat. Rev. Neurol. 2013, 9, 106–118.
- Ross, J. Science; Organization Science: Washington, DC, USA, 2022; Volume 358.
- Cable, J.; Holtzman, D.M.; Hyman, B.T.; Tansey, M.G.; Colonna, M.; Kellis, M.; Brinton, R.D.; Albert, M.; Wellington, C.L.; Sisodia, S.S.; et al. Alternatives to Amyloid for Alzheimer’s Disease Therapies—A Symposium Report. In Proceedings of the Annals of the New York Academy of Sciences; Blackwell Publishing Inc.: Hoboken, NJ, USA, 2020; Volume 1475, pp. 3–14.
- Hanseeuw, B.J.; Betensky, R.A.; Jacobs, H.I.L.; Schultz, A.P.; Sepulcre, J.; Becker, J.A.; Cosio, D.M.O.; Farrell, M.; Quiroz, Y.T.; Mormino, E.C.; et al. Association of Amyloid and Tau With Cognition in Preclinical Alzheimer Disease: A Longitudinal Study. JAMA Neurol. 2019, 76, 915.
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The Physiological Roles of Tau and Aβ: Implications for Alzheimer’s Disease Pathology and Therapeutics. Acta Neuropathol. 2020, 140, 417.
- Shimada, H.; Kitamura, S.; Shinotoh, H.; Endo, H.; Niwa, F.; Hirano, S.; Kimura, Y.; Zhang, M.R.; Kuwabara, S.; Suhara, T.; et al. Association between Aβ and Tau Accumulations and Their Influence on Clinical Features in Aging and Alzheimer’s Disease Spectrum Brains: A PBB3-PET Study. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2017, 6, 11.
- Pittman, A.M.; Fung, H.C.; de Silva, R. Untangling the Tau Gene Association with Neurodegenerative Disorders. Hum. Mol. Genet. 2006, 15, R188–R195.
- Yamada, K.; Cirrito, J.R.; Stewart, F.R.; Jiang, H.; Finn, M.B.; Holmes, B.B.; Binder, L.I.; Mandelkow, E.M.; Diamond, M.I.; Lee, V.M.Y.; et al. In Vivo Microdialysis Reveals Age-Dependent Decrease of Brain Interstitial Fluid Tau Levels in P301S Human Tau Transgenic Mice. J. Neurosci. 2011, 31, 13110–13117.
- Muralidar, S.; Ambi, S.V.; Sekaran, S.; Thirumalai, D.; Palaniappan, B. Role of Tau Protein in Alzheimer’s Disease: The Prime Pathological Player. Int. J. Biol. Macromol. 2020, 163, 1599–1617.
- Goedert, M.; Eisenberg, D.S.; Crowther, R.A. Propagation of Tau Aggregates and Neurodegeneration. Annu. Rev. Neurosci. 2017, 40, 189–210.
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Guerrero-Muñoz, M.J.; Jackson, G.R.; Kayed, R. Preparation and Characterization of Neurotoxic Tau Oligomers. Biochemistry 2010, 49, 10039–10041.
- Mattsson-Carlgren, N.; Andersson, E.; Janelidze, S.; Ossenkoppele, R.; Insel, P.; Strandberg, O.; Zetterberg, H.; Rosen, H.J.; Rabinovici, G.; Chai, X.; et al. Aβ Deposition Is Associated with Increases in Soluble and Phosphorylated Tau That Precede a Positive Tau PET in Alzheimer’s Disease. Sci. Adv. 2020, 6, eaaz2387.
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Review Interaction between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. Int. J. Biol. Sci. 2021, 17, 2181–2192.
- Small, S.A.; Duff, K. Linking Aβ and Tau in Late-Onset Alzheimer’s Disease: A Dual Pathway Hypothesis. Neuron 2008, 60, 534–542.
- Zheng, W.-H.; Bastianetto, S.; Mennicken, F.; Ma, W.; Kar, S. Amyloid L Peptide Induces Tau Phosphorylation and Loss Of Cholinergic Neurons In Rat Primary Septal Cultures. Neuroscience 2002, 115, 201–211.
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.-C.; Quinlan, M.; Wisniewski, H.M.; Bindert, L.I. Abnormal Phosphorylation of the Microtubule-Associated Protein X (Tau) in Alzheimer Cytoskeletal Pathology (Alzheimer Disease/Neurofibrillary Tangles/Paired-Helical Filaments/Microtubules). Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917.
- Silva, D.F.F.; Esteves, A.R.; Oliveira, C.R.; Cardoso, S.M. Mitochondria: The Common Upstream Driver of Amyloid-and Tau Pathology in Alzheimer´s Disease. Curr. Alzheimer Res. 2011, 8, 563.
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Dé Ric Catez, F.; Smith, C.L.; Youle, R.J. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 2001, 1, 515–525.
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired Mitochondrial Dynamics and Abnormal Interaction of Amyloid Beta with Mitochondrial Protein Drp1 in Neurons from Patients with Alzheimer’s Disease: Implications for Neuronal Damage. Hum. Mol. Genet. 2011, 20, 2495–2509.
- Iijima, K.; Gatt, A.; Iijima-Ando, K. Tau Ser262 Phosphorylation Is Critical for Aβ42-Induced Tau Toxicity in a Transgenic Drosophila Model of Alzheimer’s Disease. Hum. Mol. Genet. 2010, 19, 2947–2957.
- Akama, K.T.; van Eldik, L.J. Beta-Amyloid Stimulation of Inducible Nitric-Oxide Synthase in Astrocytes Is Interleukin-1beta- and Tumor Necrosis Factor-Alpha (TNFalpha)-Dependent, and Involves a TNFalpha Receptor-Associated Factor- and NFkappaB-Inducing Kinase-Dependent Signaling Mechanism. J. Biol. Chem. 2000, 275, 7918–7924.
- Griffin, W.S.T.; Sheng, J.G.; Roberts, G.W.; Mrak, R.E. Interleukin-1 Expression in Different Plaque Types in Alzheimer’s Disease: Significance in Plaque Evolution. J. Neuropathol. Exp. Neurol. 1995, 54, 276–281.
- Mrak, R.E.; Griffin, W.S.T. Common Inflammatory Mechanisms in Lewy Body Disease and Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2007, 66, 683–686.
- Mrak, R.E.; Sheng, J.G.; Griffin, W.S.T. Glial Cytokines in Alzheimer’s Disease: Review and Pathogenic Implications. Hum. Pathol 1995, 26, 816–823.
- Tuppo, E.E.; Arias, H.R. The Role of Inflammation in Alzheimer’s Disease. Int. J. Biochem. Cell Biol. 2005, 37, 289–305.
- Nazem, A.; Sankowski, R.; Bacher, M.; Al-Abed, Y. Rodent Models of Neuroinflammation for Alzheimer’s Disease. J. Neuroinflamm. 2015, 12, 1–15.
- Saito, T.; Saido, T.C. Neuroinflammation in Mouse Models of Alzheimer’s Disease. Clin. Exp. Neuroimmunol. 2018, 9, 211.
- Gomez-Nicola, D.; Boche, D. Post-Mortem Analysis of Neuroinflammatory Changes in Human Alzheimer’s Disease. Alzheimers Res. Ther. 2015, 7, 1–8.
- Knezevic, D.; Mizrahi, R. Molecular Imaging of Neuroinflammation in Alzheimer’s Disease and Mild Cognitive Impairment. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 80, 123–131.
- Zimmer, E.R.; Leuzy, A.; Benedet, A.L.; Breitner, J.; Gauthier, S.; Rosa-Neto, P. Tracking Neuroinflammation in Alzheimer’s Disease: The Role of Positron Emission Tomography Imaging. J. Neuroinflamm. 2014, 11, 120.
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive Innate Immune Gene Activation Accompanies Brain Aging, Increasing Vulnerability to Cognitive Decline and Neurodegeneration: A Microarray Study. J. Neuroinflamm. 2012, 9, 179.
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a Central Mechanism in Alzheimer’s Disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590.
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The Devil Is in the Details. J. Neurochem. 2016, 139, 136–153.
- Grammas, P.; Martinez, J.; Sanchez, A.; Yin, X.; Riley, J.; Gay, D.; Desobry, K.; Tripathy, D.; Luo, J.; Evola, M.; et al. A New Paradigm for the Treatment of Alzheimer’s Disease: Targeting Vascular Activation. J. Alzheimer’s Dis. 2014, 40, 619–630.
- Grammas, P. Neurovascular Dysfunction, Inflammation and Endothelial Activation: Implications for the Pathogenesis of Alzheimer’s Disease. J. Neuroinflamm. 2011, 8, 26.
- Ros-Bernal, F.; Hunot, S.; Herrero, M.T.; Parnadeau, S.; Corvol, J.C.; Lu, L.; Alvarez-Fischer, D.; Carrillo-de Sauvage, M.A.; Saurini, F.; Coussieu, C.; et al. Microglial Glucocorticoid Receptors Play a Pivotal Role in Regulating Dopaminergic Neurodegeneration in Parkinsonism. Proc. Natl. Acad. Sci. USA 2011, 108, 6632–6637.
- Herrero, M.T.; Estrada, C.; Maatouk, L.; Vyas, S. Inflammation in Parkinson’s Disease: Role of Glucocorticoids. Front. Neuroanat. 2015, 9, 32.
- Faden, A.I.; Loane, D.J. Chronic Neurodegeneration after Traumatic Brain Injury: Alzheimer Disease, Chronic Traumatic Encephalopathy, or Persistent Neuroinflammation? Neurotherapeutics 2015, 12, 143–150.
- McCombe, P.A.; Henderson, R.D. The Role of Immune and Inflammatory Mechanisms in ALS. Curr. Mol. Med. 2011, 11, 246–254.
- Haase, S.; Linker, R.A. Inflammation in Multiple Sclerosis. Ther. Adv. Neurol. Disord 2021, 14.
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a Missense Mutation in the Amyloid Precursor Protein. Lett. Nat. 1991, 349, 704–706.
- Efthymiou, A.G.; Goate, A.M. Late Onset Alzheimer’s Disease Genetics Implicates Microglial Pathways in Disease Risk. Mol. Neurodegener. 2017, 12, 1–12.
- Xie, C.; Aman, Y.; Frank, J.; Donate-Lagartos, M.J.; Gudmundsrud, R.; Čechová, K.; Shi, L.; Vyhnalek, M.; Fang, E.F. Autophagic Processes in Early- and Late-Onset Alzheimer’s Disease. Autophagy Health Dis. 2022, 287–299.
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s Disease. Nat. Rev. Dis. Prim. 2015, 1, 15056.
- Xie, J.; van Hoecke, L.; Vandenbroucke, R.E. The Impact of Systemic Inflammation on Alzheimer’s Disease Pathology. Front. Immunol. 2022, 12, 5731.
- Hawkins, B.T.; Davis, T.P. The Blood-Brain Barrier/Neurovascular Unit in Health and Disease. Pharmacol. Rev. 2005, 57, 173–185.
- Kadry, H.; Noorani, B.; Cucullo, L. A Blood–Brain Barrier Overview on Structure, Function, Impairment, and Biomarkers of Integrity. Fluids Barriers CNS 2020, 17, 1–24.
- Rhea, E.M.; Banks, W.A. Role of the Blood-Brain Barrier in Central Nervous System Insulin Resistance. Front. Neurosci. 2019, 13, 521.
- Zlokovic, B.V. Neurovascular Pathways to Neurodegeneration in Alzheimer’s Disease and Other Disorders. Nat. Rev. Neurosci. 2011, 12, 723–738.
- Wong, A.D.; Ye, M.; Levy, A.F.; Rothstein, J.D.; Bergles, D.E.; Searson, P.C. The Blood-Brain Barrier: An Engineering Perspective. Front. Neuroeng. 2013, 6, 7.
- Chowdhury, E.A.; Noorani, B.; Alqahtani, F.; Bhalerao, A.; Raut, S.; Sivandzade, F.; Cucullo, L. Understanding the Brain Uptake and Permeability of Small Molecules through the BBB: A Technical Overview. J. Cereb. Blood Flow Metab. 2021, 41, 1797–1820.
- Bernardo-Castro, S.; Sousa, J.A.; Brás, A.; Cecília, C.; Rodrigues, B.; Almendra, L.; Machado, C.; Santo, G.; Silva, F.; Ferreira, L.; et al. Pathophysiology of Blood–Brain Barrier Permeability Throughout the Different Stages of Ischemic Stroke and Its Implication on Hemorrhagic Transformation and Recovery. Front. Neurol. 2020, 11, 594672.
- Carvey, P.M.; Hendey, B.; Monahan, A.J. The Blood Brain Barrier in Neurodegenerative Disease: A Rhetorical Perspective. J. Neurochem. 2009, 111, 291.
- Pardridge, W.M. Drug Transport across the Blood–Brain Barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959.
- Yang, A.C.; Stevens, M.Y.; Chen, M.B.; Lee, D.P.; Stähli, D.; Gate, D.; Contrepois, K.; Chen, W.; Iram, T.; Zhang, L.; et al. Physiological Blood–Brain Transport Is Impaired with Age by a Shift in Transcytosis. Nature 2020, 583, 425–430.
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-Brain Barrier Breakdown in Alzheimer Disease and Other Neurodegenerative Disorders. Nat. Rev. Neurol. 2018, 14, 133–150.
- Wisniewski, H.M.; Kozlowski, P.B. Evidence for Blood-Brain Barrier Changes in Senile Dementia of the Alzheimer Type (SDAT). Ann. N. Y. Acad. Sci. 1982, 396, 119–129.
- Wisniewski, H.M.; Vorbrodt, A.W.; Wegiel, J. Amyloid Angiopathy and Blood-Brain Barrier Changes in Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 1997, 826, 161–172.
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78.
- Van De Haar, H.J.; Burgmans, S.; Jansen, J.F.A.; Van Osch, M.J.P.; Van Buchem, M.A.; Muller, M.; Hofman, P.A.M.; Verhey, F.R.J.; Backes, W.H. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 2016, 281, 527–535.
- Oikari, L.E.; Pandit, R.; Stewart, R.; Cuní-López, C.; Quek, H.; Sutharsan, R.; Rantanen, L.M.; Oksanen, M.; Lehtonen, S.; de Boer, C.M.; et al. Altered Brain Endothelial Cell Phenotype from a Familial Alzheimer Mutation and Its Potential Implications for Amyloid Clearance and Drug Delivery. Stem Cell Rep. 2020, 14, 924–939.
- Takeda, S.; Sato, N.; Ikimura, K.; Nishino, H.; Rakugi, H.; Morishita, R. Increased Blood-Brain Barrier Vulnerability to Systemic Inflammation in an Alzheimer Disease Mouse Model. Neurobiol. Aging 2013, 34, 2064–2070.
- Wang, D.; Chen, F.; Han, Z.; Yin, Z.; Ge, X.; Lei, P. Relationship Between Amyloid-β Deposition and Blood–Brain Barrier Dysfunction in Alzheimer’s Disease. Front. Cell. Neurosci. 2021, 15, 695479.
- Erickson, M.A.; Banks, W.A. Blood-Brain Barrier Dysfunction as a Cause and Consequence of Alzheimer’s Disease. J. Cereb. Blood Flow Metab. 2013, 33, 1500–1513.
- Wu, Y.C.; Sonninen, T.M.; Peltonen, S.; Koistinaho, J.; Lehtonen, Š. Blood–Brain Barrier and Neurodegenerative Diseases—Modeling with Ipsc-derived Brain Cells. Int. J. Mol. Sci. 2021, 22, 7710.
- Thomsen, M.S.; Routhe, L.J.; Moos, T. The Vascular Basement Membrane in the Healthy and Pathological Brain. J. Cereb. Blood Flow Metab. 2017, 37, 3300–3317.
- Nguyen, B.; Bix, G.; Yao, Y. Basal Lamina Changes in Neurodegenerative Disorders. Mol. Neurodegener. 2021, 16, 1–25.
- Hohsfield, L.A.; Humpel, C. Migration of Blood Cells to β-Amyloid Plaques in Alzheimer’s Disease. Exp. Gerontol. 2015, 65, 8–15.
- Pietronigro, E.; Zenaro, E.; Constantin, G. Imaging of Leukocyte Trafficking in Alzheimer’s Disease. Front. Immunol. 2016, 7, 1.
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.P.; Rivest, S. Bone Marrow-Derived Microglia Play a Critical Role in Restricting Senile Plaque Formation in Alzheimer’s Disease. Neuron 2006, 49, 489–502.
- Zenaro, E.; Pietronigro, E.; Bianca, V.d.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils Promote Alzheimer’s Disease-like Pathology and Cognitive Decline via LFA-1 Integrin. Nat. Med. 2015, 21, 880–886.
- Michaud, J.P.; Bellavance, M.A.; Préfontaine, P.; Rivest, S. Real-Time in Vivo Imaging Reveals the Ability of Monocytes to Clear Vascular Amyloid Beta. Cell Rep. 2013, 5, 646–653.
- Kook, S.Y.; Hong, H.S.; Moon, M.; Ha, C.M.; Chang, S.; Mook-Jung, I. Aβ1–42-RAGE Interaction Disrupts Tight Junctions of the Blood-Brain Barrier via Ca2+-Calcineurin Signaling. J. Neurosci. 2012, 32, 8845–8854.
- Middeldorp, J.; Lehallier, B.; Villeda, S.A.; Miedema, S.S.M.; Evans, E.; Czirr, E.; Zhang, H.; Luo, J.; Stan, T.; Mosher, K.I.; et al. Preclinical Assessment of Young Blood Plasma for Alzheimer Disease. JAMA Neurol. 2016, 73, 1325–1333.
- Sha, S.J.; Deutsch, G.K.; Tian, L.; Richardson, K.; Coburn, M.; Gaudioso, J.L.; Marcal, T.; Solomon, E.; Boumis, A.; Bet, A.; et al. Safety, Tolerability, and Feasibility of Young Plasma Infusion in the Plasma for Alzheimer Symptom Amelioration Study: A Randomized Clinical Trial. JAMA Neurol. 2019, 76, 35–40.
- De Miguel, Z.; Khoury, N.; Betley, M.J.; Lehallier, B.; Willoughby, D.; Olsson, N.; Yang, A.C.; Hahn, O.; Lu, N.; Vest, R.T.; et al. Exercise Plasma Boosts Memory and Dampens Brain Inflammation via Clusterin. Nature 2021, 600, 494–499.
- Horowitz, A.M.; Fan, X.; Bieri, G.; Smith, L.K.; Sanchez-Diaz, C.I.; Schroer, A.B.; Gontier, G.; Casaletto, K.B.; Kramer, J.H.; Williams, K.E.; et al. Blood Factors Transfer Beneficial Effects of Exercise on Neurogenesis and Cognition to the Aged Brain. Science 2020, 369, 167–173.
- Meng, Q.; Lin, M.S.; Tzeng, I.S. Relationship Between Exercise and Alzheimer’s Disease: A Narrative Literature Review. Front. Neurosci. 2020, 14, 131.
- Zhou, S.; Chen, S.; Liu, X.; Zhang, Y.; Zhao, M.; Li, W. Physical Activity Improves Cognition and Activities of Daily Living in Adults with Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Int. J. Environ. Res. Public Health 2022, 19, 1216.
- Caserta, M.T.; Caccioppo Gregory Lapin, D.D.; Ragin, A.; Groothuis, D.R. Blood-Brain Barrier Integrity in Alzheimer’s Disease Patients and Elderly Control. Subjects. J. Neuropsychiatry Clin. Neurosci. 1998, 10, 78–84.
- Schlageter, N.L.; Carson, R.E.; Rapoport, S.I. Examination of Blood—Brain Barrier Permeability in Dementia of the Alzheimer Type with EDTA and Positron Emission Tomography. J. Cereb. Blood Flow Metab. 1987, 7, 1–8.
- Starr, J.M.; Farrall, A.J.; Armitage, P.; McGurn, B.; Wardlaw, J. Blood-Brain Barrier Permeability in Alzheimer’s Disease: A Case-Control MRI Study. Psychiatry Res. Neuroimaging 2009, 171, 232–241.
- Pardridge, W.M. Treatment of Alzheimer’s Disease and Blood–Brain Barrier Drug Delivery. Pharmaceuticals 2020, 13, 1–25.