Since 1906, when Dr. Alois Alzheimer first described in a patient “a peculiar severe disease process of the cerebral cortex”, people suffering from this pathology have been waiting for a breakthrough therapy. Aducanumab, the first disease-modifying therapy (DMT) has been approved for AD, and several DMTs are in advanced stages of clinical development or regulatory review. Small molecules, mAbs, or multimodal strategies showing promise in animal studies have not confirmed that promise in the clinic (where small to moderate changes in clinical efficacy have been observed), and therefore, there is a significant unmet need for a better understanding of the AD pathogenesis and the exploration of alternative etiologies and therapeutic effective disease-modifying therapies strategies for AD.
- Alzheimer’s disease
- Aβ
- tau
- disease-modifying therapies
1. Introduction
2. Cholinergic and Dopaminergic System and Ca2+ Signaling
There is evidence that biological dysfunction or imbalance in neurotransmission, such as the cholinergic and glutamatergic systems, are a key feature in the pathophysiology of AD dementia [17]. Pathological evidence regarding AD shows that degeneration in cholinergic neuron-rich regions is associated with memory loss, agitation, and apathy [18]. Acetylcholine (ACh) has been shown to be highly correlated with memory function, including memory encoding, consolidation storage, and the retrieval process and deficits in both concentration and function of ACh have been found in AD patients. Cholinesterase inhibitors (AChEIs) can improve AD symptoms by preventing synaptic ACh breakdown in the brain. Currently, at least three AChEIs (donepezil, galantamine, and rivastigmine) are approved and being used to treat AD, with some clinical improvement in cognition and global function. However, AChEIs can only improve cognitive symptoms of AD for a certain period but cannot modify the disease course, and benefits are limited, possibly because dosing is restricted by peripheral cholinergic stimulation, which induces several side effects (dose-limiting tolerability issues, including nausea, emesis, diarrhea, muscle cramps, and general malaise). All AChEIs are approved by the Food and Drug Administration (FDA) for mild-to-moderate AD [19]. Over the course of AD, ACh levels decrease due to progressive degradation of pre-synaptic neurons that release ACh into the synaptic cleft. Hence, the efficacy of AChEI decreases with AD progression. Huperzine A, derived from the herb Huperzia serrata has a mechanism of action similar to other AChEIs and also has shown to have antioxidant and neuroprotective properties counteracting glutamate-induced toxicity [20]. A meta-analysis of Huperzine efficacy in clinical trials was inconclusive [21]. Another important therapeutic approach involves the use of drugs that act directly on the glutamatergic system, such as memantine. Glutamate is the primary excitatory neurotransmitter in the brain, acting at ionotropic and metabotropic glutamate receptors [22]. Excessive stimulation of glutamatergic signaling results in excitotoxicity, principally mediated by excessive Ca2+ entry, primarily through NMDARs causing synaptic dysfunction [23]. Memantine is a noncompetitive NMDAR-antagonist and provides symptomatic treatment of dementia inhibiting the pathological activation of NMDA receptors. Its neuroprotective effects have been demonstrated in several neurological disorders and are used for cognitive disorders in patients with moderate to severe AD. Additionally, clinical practice guidelines (EMA, NICE) and Scientific Societies recommend the combined use of memantine with AChEIs in advanced stages, being more beneficial than monotherapy with AChEIs [24,25][24][25]. Namzaric, which combines memantine and donepezil, received FDA approval in 2014. Since then, most clinical trials have accepted standard of care (SoC) with an AChEI with or without memantine at baseline [26]. Currently, there are no clear recommendations on when to suspend the administration of these drugs, therefore, the interruption of treatment in patients with the advanced disease must be guided and monitored by individualized clinical criteria. In AD diseased neurons the levels of cytosolic Ca2+ are abnormally increased [27]. Increased intracellular cytosolic Ca2+ show close relationship with mitochondrial function [28]. Growing evidence in a variety of AD models indicates that calcium dyshomeostasis drastically alters mitochondrial activity which, in turn, initiates the characteristic pathophysiology of AD, including accumulation of amyloid β-peptide (Aβ), hyperphosphorylation of Tau, synaptic dysfunction, and neuronal death. On the other hand, neurodegeneration triggered by pathological amyloid-β or Tau requires disturbed Ca2+ signaling [29]. Hence, disturbed Ca2+ signaling and AD pathophysiology constitute a vicious cycle of mutually reinforcing processes, and, therefore, therapeutic intervention to normalize Ca2+ signaling is expected to be therapeutically beneficial. REM0046127 is a small molecule that lowers Orai calcium channel activity and thereby lowers elevated cytosolic Ca2+ to physiological levels, but not below. REM0046127 was administered for the first time in humans in the Phase 1 study (NCT04672135) generally safe and well-tolerated [30]. It is predicted that REM0046127 works both as a symptomatic and DMT for AD.3. Amyloid-β Protein
It has been demonstrated that Aβ-amyloid accumulates for 30 years to reach the level typically present in mild AD dementia. Longitudinal studies such as the Australian Imaging, Biomarker, and Lifestyle (AIBL) studies were the first to use prospective analyses to identify how abnormally high levels of Aβ-amyloid (Aβ+), detected via PET were associated with a subtle but relentless decline in memory and other aspects of higher cognition in cognitively normal (CN) older adults [31,32][31][32]. Strong genetic and biochemical evidence highlights a central role of the amyloid pathway in the pathogenesis of AD [33,34][33][34]. Even though the normal function of APP is not known, it is possibly related to the regulation of neurite outgrowth, cell adhesion, and neuron migration [35]. During the last decade, several studies have described the role of Aβ as an anti-microbial peptide [36]. The amyloid cascade hypothesis is that protein misfolding of the Aβ peptide (due to reduced clearance and/or overproduction) leads to the extracellular accumulation of toxic Aβ aggregates, including β-amyloid plaques, which are the causative factor for the initiation of the neurodegenerative cascade that includes inflammation, gliosis, neuronal damage and synaptic loss [37]. Thus, it has been hypothesized that either suppression of Aβ formation or enhanced clearance will prevent neuronal dysfunction and death. In the amyloidogenic pathway [38], Aβ is generated from the APP by the sequential proteolysis at the N-terminus of the protein by the β-secretase activity (β-site APP-cleaving enzyme (BACE)) and then processed by γ-secretase complex and secreted from cells of neuronal origin via major regulation as well as a minor constitutive secretory pathway. Aβ is a normal product of cell metabolism and is present in the plasma and in cerebrospinal fluid (CSF) in healthy individuals. The cleavage position of the γ-secretase in the transmembrane domain of APP is imprecise, resulting in the production of Aβ peptides of variable length, which are readily detected in CSF and plasma. Most secreted Aβ is Aβ1–40 but a small component (5–10%) is Aβ1–42 [39], a species that is particularly important in AD. The CSF Aβ42 levels for control groups are ≈900 pg/mL and the plasma Aβ42 levels assayed with IMR are ≈15 pg/mL [40,41][40][41]. Aβ species ending at position 42 (Aβ42) aggregate more rapidly than those ending at position 40 (Aβ40) in vitro. In addition, Aβ40 is the main component of amyloid deposition occurring in cerebral amyloid angiopathy (CAA) which has a prevalence of about 50% in patients with AD [42]. It has been shown that CAA contributes to AD dementia independently of senile plaques and neurofibrillary tangles, increase the risk of intracranial hemorrhage and hemorrhagic stroke and is associated with faster rates of cognitive decline. Therefore, CAA pathology may be an important therapeutic target in AD. When these misfolded Aβ reach a critical concentration, oligomers are formed that result in protofibrils and finally culminate in mature fibrils (Figure 1) [43]. Since monomer Aβ and fibrils are in equilibrium, the deposited plaque probably acts as a reservoir for soluble Aβ, and thus eliminating the deposits would have a multifold benefit through the reduced levels of all possible toxic forms of Aβ. The level of soluble, non-fibrillar Aβ oligomers in the brain correlates strongly with the severity of the disease, suggesting that soluble oligomeric species of Aβ, rather than the fibrillary form within amyloid plaques, likely play a pivotal role in AD pathophysiology [44]. Calcium homeostasis perturbation was found to be a ubiquitous toxicity mechanism for soluble oligomers whereas no detectable effect was observed for fibrils [45]. Previous biochemical studies showed that the Aβ deposited in AD brains is heterogeneous, a process known as segmental polymorphism [46]. In a study where Aβ species were quantified, over 90% of the Aβ present ended at Aβ42, and Aβ1–42 comprised only a small fraction of the total Aβ42, indicating that most of the Aβ42 in the AD brain was truncated or modified at its amino-terminus [47]. Prominent examples of these age-modified forms of Aβ include isomerization (isoD-Aβ) and pyroglutamate formation at the N-terminal of Aβ (pGlu-Aβ, pE-Aβ) [48]. IsoD-Aβ, is the result of a chemically spontaneous and non-enzymatic reaction, while the formation of pGlu-Aβ is the consequence of N-terminal truncation by DPP4, followed by dehydration catalyzed by Glutaminyl Cyclase (QC) to form the cyclic pyroglutamate [49]. QC has strong expression in the hippocampus and cortex [50,51][50][51]. Brain regions expressing human APP without QC do not display AβpE3 plaque formation. Studies analyzing the amino-terminus of the Aβ in the AD brain showed that Aβ3(pGlu)-42 is an important component of the Aβ deposited in senile plaques of the AD brain, constituting approximately 25% of the total Aβ42 [52]. Aβ3(pGlu)-42 is highly hydrophobic, more prone to oligomerization, and has greater resistance against proteolytic degradation. When compared with full-length Aβ forms, cortical concentrations of AβpE3 correlate more strongly with cognitive impairment and pTau and appear to be more specifically linked to the disease progression [53]. Elevated QC expression in AD tissues has been reported [54], and thus it is tempting to speculate that inhibition of QC might prevent the formation of pGlu-Aβ and suppress downstream pathophysiology [55]. Moreover, AβpE3 is less prevalent in the cerebral vasculature relative to other forms of Aβ and may induce fewer instances of ARIA due to the lower prevalence of AβpE3 in the cerebral vasculature.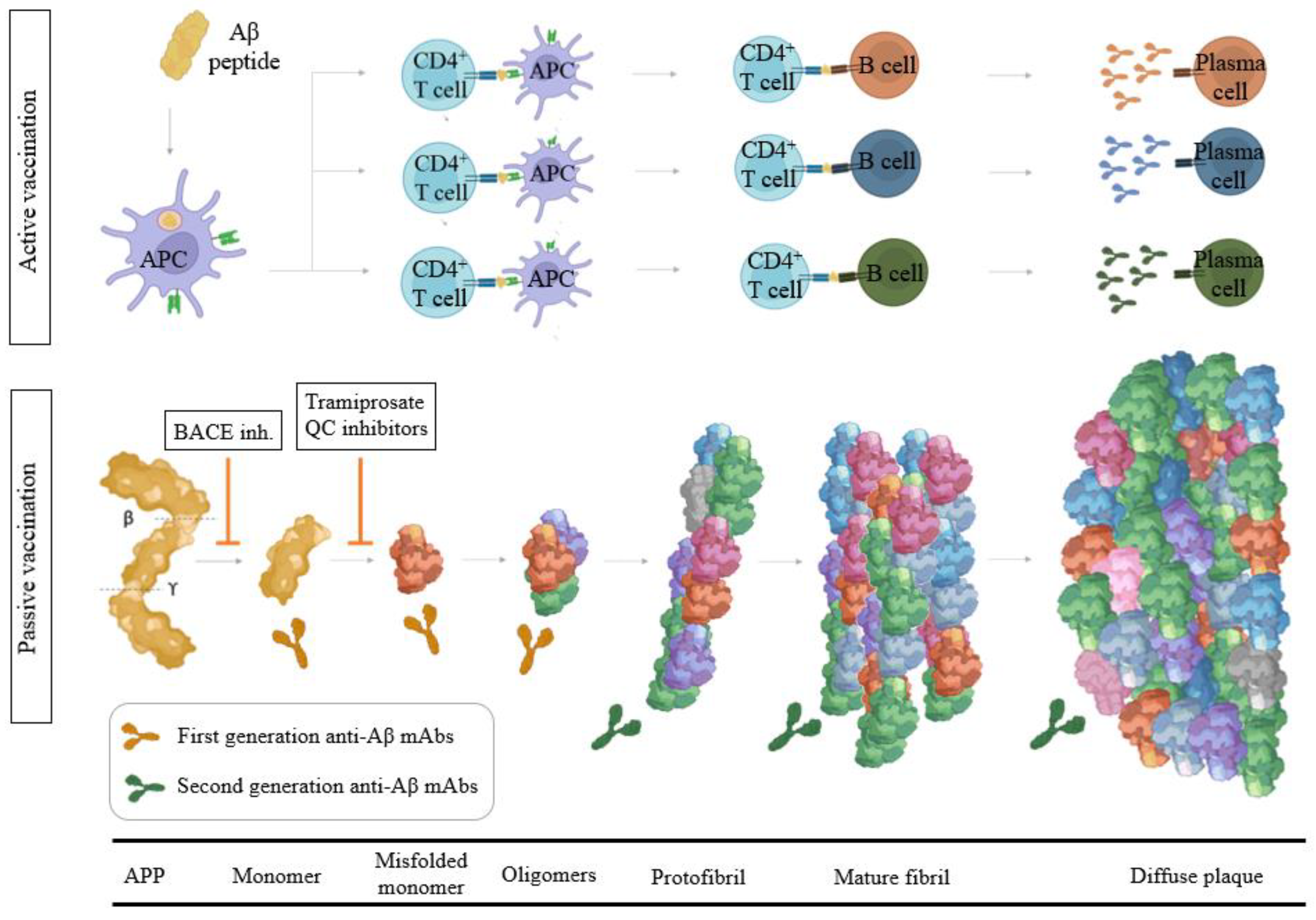
3.1. Modulators of α-, β-, and γ-Secretase Activity
Blocking the Aβ generation can be attained by blocking the enzymes involved in their production. Despite the fact that it was very appealing for many researchers to develop several inhibitors for γ-secretase, the results of its inhibition were not satisfactory. Transgenic PS1−/− mice were unhealthy, not fertile, and had lagging subventricular areas and cortical dysplasia [56]. Thus, blocking γ-secretase is more likely to result in adverse effects due to the vital biological function of PS1. BACE1 possesses structural similarities with other aspartyl proteases (such as BACE2, pepsin, renin, cathepsin D, and cathepsin E) [57]. Thus, achieving selectivity in BACE1 inhibition is crucial for developing effective BACE1 inhibitors. BACE1 inhibition provides multiple advantages, among which is the prevention of Aβ formation at an early stage of APP processing. Moreover, BACE1−/− mice exhibited a total loss of Aβ production without any significant side effects [58]. BACE1 inhibitors achieved in humans what they were designed for, namely a dose-dependent lowering of Aβ in the CNS [59]. Importantly, at the time these therapies were developed, a high level of BACE1 inhibition to achieve maximal Aβ lowering (i.e., greater than 60% lowering) was the goal. Several BACE1 inhibitors progressed into late stages of clinical trials all of which unfortunately have been terminated due to the lack of efficacy [60], toxicity (i.e., elevated liver enzymes), and dose-related cognitive worsening. The reason for the absence or even negative effects on cognition is still a matter of debate [61]. One proposed explanation has been that trials were run in too advanced AD populations and tau spreading, and neurodegeneration is already manifested at a point of no return. The umibecestat trial however was run in pre-symptomatic ApoE4 carriers, which is a genetic risk factor for AD associated with increased Aβ aggregate buildup, and still did not meet the endpoint. An explanation could be that inhibition of BACE1 interferes with the processing of the many protein substrates of BACE1, and some of these substrates are related to synaptic plasticity and synaptic homeostasis (i.e., SEZ6 and NRG1). All this clinical trial data suggest on-target toxicity is likely a contributing factor, which implies the only potential future of BACE1 inhibitors lies in a careful titration of highly selective compounds in early populations where the amyloid burden is still minimal as prophylactic therapy, or as affordable oral maintenance therapy following amyloid-clearing therapies with the goal of to achieve a low level of BACE1 inhibition (i.e., Aβ lowering 15–30%) [62]. Support for this hypothesis can be found indirectly in the protective A673T APP mutation that leads to only 25% reduced Aβ production and still avoids carriers getting AD [63]. It should be noted that these mutations lead to a reduction in Aβ production from birth onwards, which will be difficult to reproduce via pharmacological treatment.3.2. Aβ Immunotherapy
Active vaccination with Aβ1–42 was first described by Schenk et al. in 1999 [64]. Their report demonstrated that active immunization with Aβ1–42 in the PDAPP transgenic mouse reduced levels of Aβ deposits dramatically and protected mice from memory deficits. Since then, several studies have shown that both active and passive immunotherapy were effective in reducing amyloid deposition in transgenic mice models when performed as a preventative measure; however, when these approaches are performed in aged transgenic mice with pre-existing deposits, they showed diminished or no efficacy. Three main mechanisms of action for Aβ immunotherapy have been postulated: soluble equilibrium, phagocytosis, or blockade of amyloid seeding [65]. The soluble equilibrium mechanism is based upon antibodies neutralizing soluble Aβ and shifting the equilibrium to favor dissolution. This mechanism of action is proposed to take place in both the periphery and central compartments. The phagocytosis mechanism requires antibodies to gain access to the CNS, where they engage deposited amyloid and facilitate microglial-mediated phagocytosis of the plaque. Finally, others have postulated that the prevention of amyloid deposition may be due to antibodies binding to early amyloid seeds at a point in the cascade when these species are present at low abundance, thus preventing amyloid propagation.3.2.1. Aβ Passive Immunotherapy
Passive immunotherapy involves the direct administration of external antibodies. Intravenous immunoglobulin (IVIG) products were first investigated as possible therapeutic agents for MCI and AD but disappointing results were obtained in Phase 2/3 AD trials [66,67][66][67]. Aβ monoclonal antibodies (mAbs) in development target different epitopes and vary in their detection and binding affinity to several Aβ species. High doses of antibodies in the periphery are required because of the low-level (0.1–0.3%) penetration across the blood-brain barrier (BBB) to effectively engage the local (i.e., CNS) mechanisms for clearing the cerebral amyloid [68]. First-generation anti-Aβ antibody-based immunotherapy drugs were terminated in clinical trials due to the lack of cognitive benefits for AD patients (Figure 1). It is hypothesized that the inability of the N-terminal antibodies to remove existing plaque was due to antibody saturation with soluble Aβ upon entering the CNS. Bapineuzumab was administered at a maximal dose of 2 mg/kg (showing 14% ARIA-E) [69]. This mAb does not bind to truncated Aβ species as it recognizes the 1–5 of the Aβ epitope. Solanezumab, a humanized monoclonal IgG1 antibody of m266, with epitope 16–26 targeting soluble Aβ species (this epitope is buried in aggregated forms) and exhibited strong binding to monomers of Aβ40 or Aβ42 and also inhibited primary nucleation [70]. Phase 3 Expedition 3 clinical trial with solanezumab at 400 mg monthly infusions showed an inability to significantly reduce amyloid cortical burden (with no related ARIA-E or ARIA-H) [71]. In June 2017, the A4 study was initiated by quadrupling the dose from 400 to 1600 mg in symptomatic brain amyloid deposition. The trial will run until the end of 2022 (NCT02008357). Crenezumab is a humanized anti-Aβ IgG4 against the 13–24 Aβ epitope that binds monomeric and aggregated forms of Aβ, with the highest affinity for soluble oligomers, and it can block aggregation of monomers and induce disaggregation of existing Aβ aggregates in vitro. Effects were evaluated in ABBY (NCT01343966) and BLAZE (NCT01397578) Phase 2 trials with mild-AD patients were a dose up to 60–120 mg/kg were evaluated [72]. AβO levels were significantly decreased and Aβ42 monomer levels were increased in cerebrospinal fluid but PET amyloid load was not lowered [73]. Encouragingly, recent studies using second-generation mAbs that target highly specific epitopes against aggregated Aβ have shown substantial reductions in PET amyloid, with many participants becoming amyloid negative by 6–14 months of high dose treatment, showing that some indication of slowing cognitive decline can be achieved with anti-amyloid immunotherapy (Figure 1) [74]. These mAbs acts centrally recruiting microglia and also, reducing secondary nucleation. Aducanumab, (Aduhelm, BIIB037) on 7 June 2021, become the first DMT approved by the FDA for the treatment of early AD [75,76][75][76]. Aducanumab is a high-affinity, fully human IgG1 mAb against Aβ epitope 3–7 and binds to soluble Aβ aggregates and insoluble fibrils with >10,000-fold selectivity over monomers and also has demonstrated inhibition of secondary nucleation. Amyloid deposition was reduced in all treatment groups at 10 mg/kg during 26 weeks [77[77][78],78], 41.3% in the combined 10 mg/kg aducanumab group (n = 1029) experienced ARIA [79]. Lecanemab (BAN2401) is the humanized IgG1 version of the mouse mAb158, which selectively binds to large, soluble Aβ protofibrils. The antibody administered at a dose of 10 mg/kg bi-weekly reduced brain amyloid by up to 93 percent in the highest-dose group [80]. This dose slowed cognitive decline by 47 percent on the ADAS-Cog, and by 30 percent on the ADCOMS. Donanemab is a plaque-specific antibody that targets Aβp3–42, which showed rapid—a six-month course of 20 mg/kg-and robust clearance of pre-existing plaque slowing decline on the iADRS by 32 percent compared to placebo at 18 months and 27% of patients treated with the mAb developed ARIA-E [81]. Donanemab is a humanized IgG1 antibody developed from the murine IgG2a antibody mE8. Donanemab is itself strongly immunogenic and 90% of patients who received the mAb mounted an immune response against it (presence of anti-drug antibodies, ADA, which could potentially impact pharmacokinetics). Gantenerumab is a fully human IgG1 antibody designed to bind with subnanomolar affinity to Aβ fibrils binding two discontiguous regions of Aβ, with the highest affinity at residues 2–11 and 18–27 (residues 3–11 and 18–27, which are hidden within most fibrils). Two years of high-dose (1020 mg/month) subcutaneous administration of higher doses of gantenerumab in the SCarlet and Marguerite RoAD extension studies lowered brain amyloid by an average of 59 centiloid, opening the possibility of at-home administration. Novel strategies are currently under development to increase the BBB penetrance of mAbs. RG6102 is a hybrid molecule that consists of the Fc portion of gantenerumab, i.e., its tail, conjugated to the Fab shuttle that binds to transferrin. In mice, a mouse version of RG6102 entered the brain in 12-fold higher quantities than gantenerumab and cleared plaque at lower doses [82,83][82][83]. The brain shuttle-gantenerumab CSF/plasma ratio was 0.8% becoming the first evidence of a brain-shuttling effect on the CNS compartment in humans. Lecanemab (NCT03887455 and NCT04468659), Gantenerumab (NCT03444870), and Donanemab (NCT05026866) are currently under Phase 3 of clinical development and FDA has granted breakthrough therapy designation [84], and different Phase 3 head-to-head trials comparing anti-Aβ mAbs to assess superiority of brain amyloid plaque clearance are ongoing (NCT05108922). However, passive immunotherapy trials have been associated with the highly frequent occurrence of Amyloid Related Imaging Abnormalities (ARIA), referring to a spectrum of imaging abnormalities detected on MRI scans suggestive of ARIA-E or ARIA-H corresponding to microhaemorrhages and hemosiderin deposits and are associated with an accumulation of amyloid in the cerebrovasculature [85,86][85][86]. Interestingly, prolonged intracerebroventricular (icv) delivery of anti-Aβ antibodies dose-dependently reduced the parenchymal plaque burden, astrogliosis, and dystrophic neurites at doses 10- to 50-fold lower than used with systemic delivery of the same antibody in an aged Tg2576 mouse model of AD and icv-infused antibodies reduced CAA and associated micro-hemorrhages compared with systemically delivered anti-Aβ mAbs [87].3.2.2. Aβ Active Immunotherapy
Active vaccination is defined as introducing an exogenous substance to stimulate the immune system to mount a response (Figure 1) [88]. The multitargeted profile of the polyclonal antibodies generated by active vaccines may improve their probability of success in patients at different AD pathological stages with regard to single-target mAbs [89]. Active immunization with aggregated full-length Aβ1–42 (AN1792) associated with a Th1 adjuvant, was the first immunotherapy tested in AD patients, which generated anti-Aβ antibody responses in 25% of patients and showed decreased level of tau protein in the CSF and demonstrated some clinical benefit. In this trial [90], 18 patients out of 298 given the AN-1792 vaccine developed treatment-related meningoencephalitis likely caused by autoreactive T-cell activation and Aβ-reactive T-cell infiltration into the CNS and the sponsor suspended the trial [91,92][91][92]. Several second-generation Aβ-targeting vaccines have been subsequently designed to minimize Aβ-related T-cell inflammation including: ACC-001 using Aβ1–7 peptide conjugated to diphtheria toxoid protein [93], CAD106 using Aβ1–6 peptide coupled to Qb virus-like particle, V950 using multivalent Aβ1–15, and affitopes AD01 and AD02 using Aβ mimetics conjugated to KLH [94,95][94][95]. CAD106 (Generation Study 1, NCT02565511) to treat individuals with the ApoE4 allele and amyloid burden without cognitive impairment is ongoing [96]. In animals, CAD106 induced Aβ-antibody titers without activating Aβ-reactive T cells. CAD106, currently in clinical Phase 3 trial, has completed two Phase 2 trials reporting acceptable safety and evoking strong serological responses in 80% of patients [97]. ACI-24 is a liposome vaccine-based array of Aβ1–15 sequences, anchored into the surface of liposomes in such a way that the peptides adopt an aggregated β-sheet structure, forming a conformational epitope [98]. UB-311 is a mixture of 2 synthetic peptides having active UBITh® helper T-cell epitopes and B-cell epitopes from the first 14 amino acids of the N-terminus of amyloid beta (Aβ1–14) [99]. In the Phase 1 study (Study V118-AD), UB-311 elicited antibodies with specificity to the Aβ1–14 domain in all participants. ABvac40 comprises multiple repeats of Aβ33–40 using the carrier protein KLH. Unlike N-terminal end Aβ-directed antibodies, anti-C-terminal end Aβ antibodies do not bind to the unprocessed protein, preventing the accumulation of potentially toxic antigen-antibody complexes around neurons and could provide protection against N-terminally truncated Aβ peptides [100]. Indeed, 88% of the patients receiving the vaccine showed specific anti-Aβ40 antibodies that recognized monomeric, oligomeric, and insoluble (plaques) forms of the Aβ40 peptide. To date, these and related vaccines have not presented convincing Aβ brain removal or clinical efficacy data. ARIA are less frequent after active anti-Aβ immunization. The number of immunizations and antibody titer should be optimized in each case due to the different stability of the peptides used for immunization and the immunogenicity of such peptides together with the reduced immune system response in elderly population.3.3. Modulators of Aβ Toxicity
TMEM97 (recently named Sigma-2 receptor) is highly found in synapses and interacts with Aβ. The TMEM97 complex allosteric antagonist CT1812 blocked the formation of the TMEM97-Aβ complex [101]. These data obtained in experimental models of AD support a role for TMEM97 in the synaptic binding of Aβ in AD where it may mediate synaptotoxicity through the modulation of intracellular Ca2+ levels. CT1812 is neuroprotective and reduces cognitive deficits and neuroinflammation [102]. Tramiprosate, and its prodrug ALZ-801 showed significant clinical effects in the homozygous for ApoE4 [103]. A novel multi-ligand enveloping stabilizing effect of the small molecule Tramiprosate, modulates conformational flexibility of Aβ and stabilizes Aβ42 monomers resulting in the prevention of Aβ-oligomer formation [104]. Reductions in p-Tau levels and dose-dependent preservation of hippocampal volume have been observed in the ApoE 4/4 population and a Phase 3 in this population is currently ongoing (NCT04770220) [105]. Pharmacological QC inhibition reduces pGlu-Aβ levels and cerebral amyloid burden, improving cognitive function in transgenic AD mouse models. To date, the only QC inhibitor in clinical trials is PQ912 [106]. Recently, a randomized, double-blind, placebo-controlled Phase 2a SAPHIR trial to evaluate doses of PQ912 for 3 months in MCI or early AD showed significantly reduced YKL-40 and neurogranin compared with the placebo group, brain electrical rhythms were normalized and benefits on working memory were observed [107]. QC activity is responsible for the conversion of monocyte chemotactic proteins to their bioactive pGlu-modified forms, and inhibition by the QC inhibitor PBD150 reduces monocyte migration. Thus, the contribution of QC activity to AD pathology may therefore be multi-faceted. Intriguingly, PBD150 reduces pGlu-Aβ levels and total amyloid burden in the brains of transgenic AD mouse models, despite its reported inability to cross the murine BBB [108,109][108][109]. A blockade of AβpE3 formation by inhibiting QC may cause uncertain side effects, given that QC can process the N-terminal of many other subtracts.References
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; Van Der Flier, W.M. Alzheimer’ s disease. Lancet 2021, 397, 1577–1590.
- GBD 2019 Dementia Forecasting Collaborators. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet 2022, 7, 105–125.
- Ávila-Villanueva, M.; Fernández-Blázquez, M.A. Subjective cognitive decline as a preclinical marker for Alzheimer’s disease: The challenge of stability over time. Front. Aging Neurosci. 2017, 9, 377.
- Aisen, P.S.; Cummings, J.; Jack, C.R.; Morris, J.C.; Sperling, R.; Frölich, L.; Jones, R.W.; Dowsett, S.A.; Matthews, B.R.; Raskin, J.; et al. On the path to 2025: Understanding the Alzheimer’s disease continuum. Alzheimer’s Res. Ther. 2017, 9, 60.
- Mendez, M.F. Early-onset Alzheimer disease and its variants. Contin. Lifelong Learn. Neurol. 2019, 25, 34–51.
- Herrera-Rivero, M. Late-Onset Alzheimer’s Disease: Risk Factors, Clinical Diagnosis and the Search for Biomarkers. Neurodegener. Dis. 2013.
- Holstege, H.; Hulsman, M.; Charbonnier, C.; Grenier-Boley, B.; Quenez, O.; Grozeva, D.; van Rooij, J.G.J.; Sims, R.; Ahmad, S.; Amin, N.; et al. Exome sequencing identifies rare damaging variants in the ATB8B4 and ABCA1 genes as novel risk factors for Alzheimer’s disease. Alzheimers. Dement. 2021, 17, e055982.
- Griciuc, A.; Tanzi, R.E. The role of innate immune genes in Alzheimer’s disease. Curr. Opin. Neurol. 2021, 34, 228–236.
- Liu, Y.; Thalamuthu, A.; Mather, K.A.; Crawford, J.; Ulanova, M.; Wong, M.W.K.; Pickford, R.; Sachdev, P.S.; Braidy, N. Plasma lipidome is dysregulated in Alzheimer’s disease and is associated with disease risk genes. Transl. Psychiatry 2021, 11, 344.
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430.
- Prokopenko, D.; Hecker, J.; Kirchner, R.; Chapman, B.A.; Hoffman, O.; Mullin, K.; Hide, W.; Bertram, L.; Laird, N.; DeMeo, D.L.; et al. Identification of Novel Alzheimer’s Disease Loci Using Sex-Specific Family-Based Association Analysis of Whole-Genome Sequence Data. Sci. Rep. 2020, 10, 5029.
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimery’s disease pathology is associated with early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014, 17, 1156–1163.
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32.
- Saito, S.; Ihara, M. Interaction between cerebrovascular disease and Alzheimer pathology. Curr. Opin. Psychiatry 2016, 29, 168–173.
- Geldmacher, D.S. Treatment guidelines for Alzheimer’s disease: Redefining perceptions in primary care. Prim. Care Companion J. Clin. Psychiatry 2007, 9, 113–121.
- Cummings, J.; Fox, N. Defining Disease Modifying Therapy for Alzheimer’S Disease. J. Prev. Alzheimer’s Dis. 2017, 4, 109.
- Snowden, S.G.; Ebshiana, A.A.; Hye, A.; Pletnikova, O.; O’Brien, R.; Yang, A.; Troncoso, J.; Legido-Quigley, C.; Thambisetty, M. Neurotransmitter Imbalance in the Brain and Alzheimer’s Disease Pathology. J. Alzheimer’s Dis. 2019, 72, 35–43.
- Ferreira-Vieira, T.H.; Isabella, M.; Guimaraes, F.R.S. Alzheimer’s Disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115.
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933.
- Friedli, M.J.; Inestrosa, N.C. Huperzine a and its neuroprotective molecular signaling in alzheimer’s disease. Molecules 2021, 26, 6531.
- Yang, G.; Wang, Y.; Tian, J.; Liu, J.P. Huperzine A for Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. PLoS ONE 2013, 8, e74916.
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2004, 45, 583–595.
- Wang, R.; Reddy, H. Role of Glutamate and NMDA in Alzheimer’s desease. J. Alzheimer’s Desese 2017, 57, 1041–1048.
- Koola, M.M. Galantamine-Memantine combination in the treatment of Alzheimer’s disease and beyond. Psychiatry Res. 2020, 293, 113409.
- Guo, J.; Wang, Z.; Liu, R.; Huang, Y.; Zhang, N.; Zhang, R. Memantine, Donepezil, or Combination Therapy—What is the best therapy for Alzheimer’s Disease? A Network Meta-Analysis. Brain Behav. 2020, 10, e01831.
- Teipel, S.; Gustafson, D.; Ossenkoppele, R.; Hansson, O.; Babiloni, C.; Wagner, M.; Riedel-Heller, S.; Kilimann, I.; Tang, Y. Alzheimer’s disease—Standard of diagnosis, treatment, care, and prevention. J. Nucl. Med. 2022, 63, 981–985.
- Woods, N.K.; Padmanabhan, J. Neuronal calcium signaling and Alzheimer’s disease. Adv. Exp. Med. Biol. 2012, 740, 1193–1217.
- Ryan, K.C.; Ashkavand, Z.; Norman, K.R. The role of mitochondrial calcium homeostasis in alzheimer’s and related diseases. Int. J. Mol. Sci. 2020, 21, 9153.
- Guan, P.P.; Cao, L.L.; Wang, P. Elevating the levels of calcium ions exacerbate alzheimer’s disease via inducing the production and aggregation of β-amyloid protein and phosphorylated tau. Int. J. Mol. Sci. 2021, 22, 5900.
- Cummings, J.; Lee, G.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s disease drug development pipeline: 2021. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2021, 7, e12179.
- van der Kall, L.M.; Truong, T.; Burnham, S.C.; Doré, V.; Mulligan, R.S.; Bozinovski, S.; Lamb, F.; Bourgeat, P.; Fripp, J.; Schultz, S.; et al. Association of β-Amyloid Level, Clinical Progression, and Longitudinal Cognitive Change in Normal Older Individuals. Neurology 2021, 96, e662–e670.
- Fowler, C.; Rainey-Smith, S.R.; Bird, S.; Bomke, J.; Bourgeat, P.; Brown, B.M.; Burnham, S.C.; Bush, A.I.; Chadunow, C.; Collins, S.; et al. Fifteen Years of the Australian Imaging, Biomarkers and Lifestyle (AIBL) Study: Progress and Observations from 2359 Older Adults Spanning the Spectrum from Cognitive Normality to Alzheimer’s Disease. J. Alzheimer’s Dis. Rep. 2021, 5, 443–468.
- Kokjohn, T.A.; Roher, A.E. Amyloid precursor protein transgenic mouse models and Alzheimer’s disease: Understanding the paradigms, limitations and contributions. Alzheimers Dement. 2009, 5, 340–347.
- Esquerda-Canals, G.; Montoliu-Gaya, L.; Güell-Bosch, J.; Villegas, S. Mouse Models of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1171–1183.
- Karisetty, B.C.; Bhatnagar, A.; Armour, E.M.; Beaver, M.; Zhang, H.; Elefant, F. Amyloid-β Peptide Impact on Synaptic Function and Neuroepigenetic Gene Control Reveal New Therapeutic Strategies for Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 577622.
- Gosztyla, M.L.; Brothers, H.M.; Robinson, S.R. Alzheimer’s Amyloid-β is an Antimicrobial Peptide: A Review of the Evidence. J. Alzheimer’s Dis. 2018, 62, 1495–1506.
- Ricciarelli, R.; Fedele, E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr. Neuropharmacol. 2017, 15, 926–935.
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503.
- Baumketner, A. Amyloid beta-protein monomer structure: A computational and experimental study. Protein Sci. 2006, 15, 420–428.
- Orellana, A.; García-González, P.; Valero, S.; Montrreal, L.; de Rojas, I.; Hernández, I.; Rosende-Roca, M.; Vargas, L.; Tartari, J.P.; Esteban-De Antonio, E.; et al. Establishing In-House Cutoffs of CSF Alzheimer’s Disease Biomarkers for the AT(N) Stratification of the Alzheimer Center Barcelona Cohort. Int. J. Mol. Sci. 2022, 23, 6891.
- Teunissen, C.E.; Chiu, M.J.; Yang, C.C.; Yang, S.Y.; Scheltens, P.; Zetterberg, H.; Blennow, K. Plasma Amyloid-β (Aβ42) Correlates with Cerebrospinal Fluid Aβ42 in Alzheimer’s Disease. J. Alzheimers. Dis. 2018, 62, 1857–1863.
- Jäkel, L.; De Kort, A.M.; Klijn, C.J.M.; Schreuder, F.H.B.M.; Verbeek, M.M. Prevalence of cerebral amyloid angiopathy: A systematic review and meta-analysis. Alzheimer’s Dement. 2022, 18, 10–28.
- Seeman, P.; Seeman, N. Alzheimer’s disease: β-amyloid plaque formation in human brain. Synapse 2011, 65, 1289–1297.
- Jongbloed, W.; Bruggink, K.A.; Kester, M.I.; Visser, P.J.; Scheltens, P.; Blankenstein, M.A.; Verbeek, M.M.; Teunissen, C.E.; Veerhuis, R. Amyloid-β oligomers relate to cognitive decline in alzheimer’s disease. J. Alzheimer’s Dis. 2015, 45, 35–43.
- Arbel-Ornath, M.; Hudry, E.; Boivin, J.R.; Hashimoto, T.; Takeda, S.; Kuchibhotla, K.V.; Hou, S.; Lattarulo, C.R.; Belcher, A.M.; Shakerdge, N.; et al. Soluble oligomeric amyloid-β induces calcium dyshomeostasis that precedes synapse loss in the living mouse brain. Mol. Neurodegener. 2017, 12, 27.
- Zampar, S.; Klafki, H.W.; Sritharen, K.; Wiltfang, T.A.B.J.; Rostagno, A.; Ghiso, J.; Miles, L.A.; Wirths, O. N-terminal heterogeneity of parenchymal and vascular amyloid-β deposits in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2020, 46, 673–685.
- Gravina, S.A.; Ho, L.; Eckman, C.B.; Long, K.E.; Otvos, L.; Younkin, L.H.; Suzuki, N.; Younkin, S.G. Amyloid β protein (Aβ) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at Aβ40 or Aβ42(43). J. Biol. Chem. 1995, 270, 7013–7016.
- Moro, M.L.; Phillips, A.S.; Gaimster, K.; Paul, C.; Mudher, A.; Nicoll, J.A.R.; Boche, D. Pyroglutamate and Isoaspartate modified Amyloid-Beta in ageing and Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 3.
- Antonyan, A.; Schlenzig, D.; Schilling, S.; Naumann, M.; Sharoyan, S.; Mardanyan, S.; Demuth, H.U. Concerted action of dipeptidyl peptidase IV and glutaminyl cyclase results in formation of pyroglutamate-modified amyloid peptides in vitro. Neurochem. Int. 2018, 113, 112–119.
- Hartlage-Rübsamen, M.; Morawski, M.; Waniek, A.; Jäger, C.; Zeitschel, U.; Koch, B.; Cynis, H.; Schilling, S.; Schliebs, R.; Demuth, H.U.; et al. Glutaminyl cyclase contributes to the formation of focal and diffuse pyroglutamate (pGlu)-Aβ deposits in hippocampus via distinct cellular mechanisms. Acta Neuropathol. 2011, 121, 705–719.
- Morawski, M.; Schilling, S.; Kreuzberger, M.; Waniek, A.; Jäger, C.; Koch, B.; Cynis, H.; Kehlen, A.; Arendt, T.; Hartlage-Rübsamen, M.; et al. Glutaminyl cyclase in human cortex: Correlation with (pGlu)-amyloid-β load and cognitive decline in Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 39, 385–400.
- Drummond, E.; Kavanagh, T.; Pires, G.; Marta-Ariza, M.; Kanshin, E.; Nayak, S.; Faustin, A.; Berdah, V.; Ueberheide, B.; Wisniewski, T. The amyloid plaque proteome in early onset Alzheimer’s disease and Down syndrome. Acta Neuropathol. Commun. 2022, 10, 53.
- Neddens, J.; Daurer, M.; Flunkert, S.; Beutl, K.; Loeffler, T.; Walker, L.; Attems, J.; Hutter-Paier, B. Correlation of pyroglutamate amyloid β and ptau Ser202/Thr205 levels in Alzheimer’s disease and related murine models. PLoS ONE 2020, 15, e0235543.
- Gunn, A.P.; Wong, B.X.; McLean, C.; Fowler, C.; Barnard, P.J.; Duce, J.A.; Roberts, B.R. Increased glutaminyl cyclase activity in brains of Alzheimer’s disease individuals. J. Neurochem. 2021, 156, 979–987.
- Sofola-Adesakin, O.; Khericha, M.; Snoeren, I.; Tsuda, L.; Partridge, L. pGluAβ increases accumulation of Aβ and exacerbates toxicity. Acta Neuropathol. Commun. 2016, 4, 109.
- Elder, G.A.; Sosa, M.A.G.; De Gasperi, R.; Dickstein, D.L.; Hof, P.R. Presenilin transgenic mice as models of Alzheimer’s disease. Brain Struct. Funct. 2010, 214, 127–143.
- Grüninger-Leitch, F.; Schlatter, D.; Küng, E.; Nelböck, P.; Döbeli, H. Substrate and inhibitor profile of BACE (β-secretase) and comparison with other mammalian aspartic proteases. J. Biol. Chem. 2002, 277, 4687–4693.
- Luo, Y.; Bolon, B.; Damore, M.A.; Fitzpatrick, D.; Liu, H.; Zhang, J.; Yan, Q.; Vassar, R.; Citron, M. BACE1 (β-secretase) knockout mice do not acquire compensatory gene expression changes or develop neural lesions over time. Neurobiol. Dis. 2003, 14, 81–88.
- Yan, R.; Robert Vassar, P. Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 2014, 13, 319–329.
- Vassar, R. BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease. Alzheimer’s Res. Ther. 2014, 6, 89.
- Lahiri, D.K.; Maloney, B.; Long, J.M.; Greig, N.H. Lessons from a BACE1 inhibitor trial: Off-site but not off base. Alzheimer’s Dement. 2014, 10, S411–S419.
- Willis, B.A.; Lowe, S.L.; Monk, S.A.; Cocke, P.J.; Aluise, C.D.; Boggs, L.N.; Borders, A.R.; Brier, R.A.; Dean, R.A.; Green, S.J.; et al. Robust Pharmacodynamic Effect of LY3202626, a Central Nervous System Penetrant, Low Dose BACE1 Inhibitor, in Humans and Nonclinical Species. J. Alzheimer’s Dis. Reports 2022, 6, 1–15.
- Xia, Q.; Yang, X.Y.; Shi, J.B.; Liu, Z.J.; Peng, Y.H.; Wang, W.J.; Li, B.W.; Zhao, Y.; Xiao, J.Y.; Huang, L.; et al. The Protective A673T Mutation of Amyloid Precursor Protein (APP) in Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 4038–4050.
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guldo, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Khan, K.; et al. Immunization with amyloid-β attenuates Alzheimer disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177.
- Menendez-Gonzalez, M.; Perez-Pinera, P.; Martinez-Rivera, M.; Lopez Muniz, A.; Vega, J.A. Immunotherapy for Alzheimer’s Disease: Rational Basis in Ongoing Clinical Trials. Curr. Pharm. Des. 2011, 17, 508–520.
- Relkin, N.R.; Thomas, R.G.; Rissman, R.A.; Brewer, J.B.; Rafii, M.S.; Van Dyck, C.H.; Jack, C.R.; Sano, M.; Knopman, D.S.; Raman, R.; et al. A phase 3 trial of IV immunoglobulin for Alzheimer disease. Neurology 2017, 88, 1768–1775.
- Kile, S.; Au, W.; Parise, C.; Rose, K.; Donnel, T.; Hankins, A.; Chan, M.; Ghassemi, A. IVIG treatment of mild cognitive impairment due to Alzheimer’s disease: A randomised double-blinded exploratory study of the effect on brain atrophy, cognition and conversion to dementia. J. Neurol. Neurosurg. Psychiatry 2017, 88, 106–112.
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972.
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two Phase 3 Trials of Bapineuzumab in Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2014, 370, 322–333.
- Crespi, G.A.N.; Hermans, S.J.; Parker, M.W.; Miles, L.A. Molecular basis for mid-region amyloid-β capture by leading Alzheimer’s disease immunotherapies. Sci. Rep. 2015, 5, 2–6.
- Willis, B.A.; Sundell, K.; Lachno, D.R.; Ferguson-Sells, L.R.; Case, M.G.; Holdridge, K.; DeMattos, R.B.; Raskin, J.; Siemers, E.R.; Dean, R.A. Central pharmacodynamic activity of solanezumab in mild Alzheimer’s disease dementia. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 652–660.
- Yoshida, K.; Moein, A.; Bittner, T.; Ostrowitzki, S.; Lin, H.; Honigberg, L.; Jin, J.Y.; Quartino, A. Pharmacokinetics and pharmacodynamic effect of crenezumab on plasma and cerebrospinal fluid beta-amyloid in patients with mild-to-moderate Alzheimer’s disease. Alzheimer’s Res. Ther. 2020, 12, 16.
- Yang, T.; Dang, Y.; Ostaszewski, B.; Mengel, D.; Steffen, V.; Rabe, C.; Bittner, T.; Walsh, D.M.; Selkoe, D.J. Target engagement in an alzheimer trial: Crenezumab lowers amyloid β oligomers in cerebrospinal fluid. Ann. Neurol. 2019, 86, 215–224.
- Tolar, M.; Abushakra, S.; Hey, J.A.; Porsteinsson, A.; Sabbagh, M. Aducanumab, gantenerumab, BAN2401, and ALZ-801—The first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimer’s Res. Ther. 2020, 12, 95.
- Yang, P.; Sun, F. Aducanumab: The first targeted Alzheimer’s therapy. Drug Discov. Ther. 2021, 15, 166–168.
- Cummings, J.; Rabinovici, G.D.; Atri, A.; Aisen, P.; Apostolova, L.G.; Hendrix, S.; Sabbagh, M.; Selkoe, D.; Weiner, M.; Salloway, S. Aducanumab: Appropriate Use Recommendations Update. J. Prev. Alzheimer’s Dis. 2022, 9, 221–230.
- Knopman, D.S.; Jones, D.T.; Greicius, M.D. Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimer’s Dement. 2021, 17, 696–701.
- Cummings, J.; Aisen, P.; Lemere, C.; Atri, A.; Sabbagh, M.; Salloway, S. Aducanumab produced a clinically meaningful benefit in association with amyloid lowering. Alzheimer’s Res. Ther. 2021, 13, 10–12.
- VandeVrede, L.; Gibbs, D.M.; Koestler, M.; La Joie, R.; Ljubenkov, P.A.; Provost, K.; Soleimani-Meigooni, D.; Strom, A.; Tsoy, E.; Rabinovici, G.D.; et al. Symptomatic amyloid-related imaging abnormalities in an APOE ε4/ε4 patient treated with aducanumab. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2020, 12, e12101.
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimer’s Res. Ther. 2021, 13, 80.
- Lowe, S.L.; Duggan Evans, C.; Shcherbinin, S.; Cheng, Y.J.; Willis, B.A.; Gueorguieva, I.; Lo, A.C.; Fleisher, A.S.; Dage, J.L.; Ardayfio, P.; et al. Donanemab (LY3002813) Phase 1b Study in Alzheimer’s Disease: Rapid and Sustained Reduction of Brain Amyloid Measured by Florbetapir F18 Imaging. J. Prev. Alzheimer’s Dis. 2021, 8, 414–424.
- Hultqvist, G.; Syvänen, S.; Fang, X.T.; Lannfelt, L.; Sehlin, D. Bivalent brain shuttle increases antibody uptake by monovalent binding to the transferrin receptor. Theranostics 2017, 7, 308–318.
- Weber, F.; Bohrmann, B.; Niewoehner, J.; Fischer, J.A.A.; Rueger, P.; Tiefenthaler, G.; Moelleken, J.; Bujotzek, A.; Brady, K.; Singer, T.; et al. Brain Shuttle Antibody for Alzheimer’s Disease with Attenuated Peripheral Effector Function due to an Inverted Binding Mode. Cell Rep. 2018, 22, 149–162.
- Tian Hui Kwan, A.; Arfaie, S.; Therriault, J.; Rosa-Neto, P.; Gauthier, S. Lessons Learnt from the Second Generation of Anti-Amyloid Monoclonal Antibodies Clinical Trials. Dement. Geriatr. Cogn. Disord. 2021, 49, 334–348.
- Withington, C.G.; Turner, R.S. Amyloid-Related Imaging Abnormalities With Anti-amyloid Antibodies for the Treatment of Dementia Due to Alzheimer’s Disease. Front. Neurol. 2022, 13, 862369.
- Sperling, R.A.; Jack, C.R.; Black, S.E.; Frosch, M.P.; Greenberg, S.M.; Hyman, B.T.; Scheltens, P.; Carrillo, M.C.; Thies, W.; Bednar, M.M.; et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: Recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimer’s Dement. 2011, 7, 367–385.
- Chouhan, N.B.; Siegel, G.J. Intracerebroventricular passive immunization in transgenic mouse models of Alzheimer’s disease. Expert Rev. Vaccines 2004, 3, 717–725.
- Cacabelos, R. How plausible is an Alzheimer’s disease vaccine? Expert Opin. Drug Discov. 2020, 15, 1–6.
- Asuni, A.A.; Boutajangout, A.; Scholtzova, H.; Knudsen, E.; Li, Y.S.; Quartermain, D.; Frangione, B.; Wisniewski, T.; Sigurdsson, E.M. Vaccination of Alzheimer’s model mice with Aβ derivative in alum adjuvant reduces Aβ burden without microhemorrhages. Eur. J. Neurosci. 2006, 24, 2530–2542.
- Gilman, S.; Koller, M.; Black, R.S.; Jenkins, L.; Griffith, S.G.; Fox, N.C.; Eisner, L.; Kirby, L.; Rovira, M.B.; Forette, F.; et al. Clinical Effects of Abeta immunization(AN1792) in patients with AD in an interrupted trial. Neurology 2005, 64, 1553–1562.
- Fox, N.C.; Black, R.S.; Gilman, S.; Rossor, M.N.; Griffith, S.G.; Jenkins, L.; Koller, M. Effects of Aβ immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology 2005, 64, 1563–1572.
- Vellas, B.; Black, R.; Thal, L.; Fox, N.; Daniels, M.; McLennan, G.; Tompkins, C.; Leibman, C.; Pomfret, M.; Grundman, M. Long-Term Follow-Up of Patients Immunized with AN1792: Reduced Functional Decline in Antibody Responders. Curr. Alzheimer Res. 2009, 6, 144–151.
- Hull, M.; Sadowsky, C.; Arai, H.; Leterme, G.L.P.; Holstein, A.; Booth, K.; Peng, Y.; Yoshiyama, T.; Suzuki, H.; Ketter, N.; et al. Long-Term Extensions of Randomized Vaccination Trials of ACC-001 and QS-21 in Mild to Moderate Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 696–708.
- Mandler, M.; Santic, R.; Gruber, P.; Cinar, Y.; Pichler, D.; Funke, S.A.; Willbold, D.; Schneeberger, A.; Schmidt, W.; Mattner, F. Tailoring the antibody response to aggregated aβ using novel Alzheimer-vaccines. PLoS ONE 2015, 10, e0115237.
- Vassilakopoulou, V.; Karachaliou, C.E.; Evangelou, A.; Zikos, C.; Livaniou, E. Peptide-based vaccines for neurodegenerative diseases: Recent endeavors and future perspectives. Vaccines 2021, 9, 1278.
- Winblad, B.; Andreasen, N.; Minthon, L.; Floesser, A.; Imbert, G.; Dumortier, T.; Maguire, R.P.; Blennow, K.; Lundmark, J.; Staufenbiel, M.; et al. Safety, tolerability, and antibody response of active Aβ immunotherapy with CAD106 in patients with Alzheimer’s disease: Randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 2012, 11, 597–604.
- Vandenberghe, R.; Riviere, M.E.; Caputo, A.; Sovago, J.; Maguire, R.P.; Farlow, M.; Marotta, G.; Sanchez-Valle, R.; Scheltens, P.; Ryan, J.M.; et al. Active Aβ immunotherapy CAD106 in Alzheimer’s disease: A phase 2b study. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 10–22.
- Muhs, A.; Hickman, D.T.; Pihlgren, M.; Chuard, N.; Giriens, V.; Meerschman, C.; Van Der Auwera, I.; Van Leuven, F.; Sugawara, M.; Weingertner, M.C.; et al. Liposomal vaccines with conformation-specific amyloid peptide antigens define immune response and efficacy in APP transgenic mice. Proc. Natl. Acad. Sci. USA 2007, 104, 9810–9815.
- Wang, C.Y.; Wang, P.N.; Chiu, M.J.; Finstad, C.L.; Lin, F.; Lynn, S.; Tai, Y.H.; De Fang, X.; Zhao, K.; Hung, C.H.; et al. UB-311, a novel UBITh® amyloid β peptide vaccine for mild Alzheimer’s disease. Alzheimer’s Dement. 2017, 3, 262–272.
- Lacosta, A.M.; Pascual-Lucas, M.; Pesini, P.; Casabona, D.; Pérez-Grijalba, V.; Marcos-Campos, I.; Sarasa, L.; Canudas, J.; Badi, H.; Monleón, I.; et al. Safety, tolerability and immunogenicity of an active anti-Aβ 40 vaccine (ABvac40) in patients with Alzheimer’s disease: A randomised, double-blind, placebo-controlled, phase i trial. Alzheimer’s Res. Ther. 2018, 10, 12.
- Grundman, M.; Morgan, R.; Lickliter, J.D.; Schneider, L.S.; DeKosky, S.; Izzo, N.J.; Guttendorf, R.; Higgin, M.; Pribyl, J.; Mozzoni, K.; et al. A phase 1 clinical trial of the sigma-2 receptor complex allosteric antagonist CT1812, a novel therapeutic candidate for Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 20–26.
- Izzo, N.J.; Yuede, C.M.; LaBarbera, K.M.; Limegrover, C.S.; Rehak, C.; Yurko, R.; Waybright, L.; Look, G.; Rishton, G.; Safferstein, H.; et al. Preclinical and clinical biomarker studies of CT1812: A novel approach to Alzheimer’s disease modification. Alzheimer’s Dement. 2021, 17, 1365–1382.
- Hey, J.A.; Kocis, P.; Hort, J.; Abushakra, S.; Power, A.; Vyhnálek, M.; Yu, J.Y.; Tolar, M. Discovery and Identification of an Endogenous Metabolite of Tramiprosate and Its Prodrug ALZ-801 that Inhibits Beta Amyloid Oligomer Formation in the Human Brain. CNS Drugs 2018, 32, 849–861.
- Manzano, S.; Agüera, L.; Aguilar, M.; Olazarán, J. A Review on Tramiprosate (Homotaurine) in Alzheimer’s Disease and Other Neurocognitive Disorders. Front. Neurol. 2020, 11, 614.
- Hey, J.A.; Yu, J.Y.; Versavel, M.; Abushakra, S.; Kocis, P.; Power, A.; Kaplan, P.L.; Amedio, J.; Tolar, M. Clinical Pharmacokinetics and Safety of ALZ-801, a Novel Prodrug of Tramiprosate in Development for the Treatment of Alzheimer’s Disease. Clin. Pharmacokinet. 2018, 57, 315–333.
- Scheltens, P.; Hallikainen, M.; Grimmer, T.; Duning, T.; Gouw, A.A.; Teunissen, C.E.; Wink, A.M.; Maruff, P.; Harrison, J.; Van Baal, C.M.; et al. Safety, tolerability and efficacy of the glutaminyl cyclase inhibitor PQ912 in Alzheimer’s disease: Results of a randomized, double-blind, placebo-controlled phase 2a study. Alzheimer’s Res. Ther. 2018, 10, 107.
- Vijverberg, E.G.B.; Axelsen, T.M.; Bihlet, A.R.; Henriksen, K.; Weber, F.; Fuchs, K.; Harrison, J.E.; Kühn-Wache, K.; Alexandersen, P.; Prins, N.D.; et al. Rationale and study design of a randomized, placebo-controlled, double-blind phase 2b trial to evaluate efficacy, safety, and tolerability of an oral glutaminyl cyclase inhibitor varoglutamstat (PQ912) in study participants with MCI and mild AD—VIVIAD. Alzheimer’s Res. Ther. 2021, 13, 142.
- Cynis, H.; Scheel, E.; Saido, T.C.; Schilling, S.; Demuth, H.U. Amyloidogenic processing of amyloid precursor protein: Evidence of a pivotal role of glutaminyl cyclase in generation of pyroglutamate-modified amyloid-β. Biochemistry 2008, 47, 7405–7413.
- Brooksa, A.F.; Jacksona, I.M.; Shaoa, X.; Kropoga, G.W.; Shermana, P.; Quesadaa, C.A.; Scotta, P.J.H. Synthesis and evaluation of PBD150, a radiolabeled glutaminyl cyclase inhibitor for the potential detection of Alzheimer’s disease prior to amyloid β aggregation. Medchemcomm 2015, 1, 1065–1068.