Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jessie Wu and Version 3 by Jessie Wu.
Spinocerebellar ataxias (SCAs) are a heterogeneous group of autosomal dominantly inherited progressive disorders with degeneration and dysfunction of the cerebellum. The genetic background of SCAs can be classified into two groups: Group I representing repeat expansion SCAs, such as SCA1 and SCA2 which are caused by dynamic repeat expansion mutations, typically polyglutamine repeat expansions, and Group II representing conventional mutation SCAs (non-repeat expansion SCAs), which are caused by nonsense mutations, missense mutations, deletions or insertions, such as SCA5 or SCA14.
- mGluR1
- RGS8
- spinocerebellar ataxias
1. Abnormalities of mGluR1 Signaling Associated with Pathogenesis of Spinocerebellar Ataxias
Based on the global transcriptional data, several dysregulated molecules have been found from different mouse models with cerebellar ataxic phenotype, indicating potentially important pathways associated with pathogenesis of SCAs and cerebellar ataxia. For example, staggerer mice exhibit a characteristic severe cerebellar ataxia due to an underdeveloped cerebellar cortex and unaligned Purkinje cells [1]. Overlap based analysis of microarray data from the SCA1 mouse model [2] and staggerer mouse [3] was performed and mGluR1 was identified as a common molecule from overlapping the data of both mouse models. This molecule and associated signaling will provide a better understanding of the disease mechanisms of SCAs and cerebellar ataxia.
A variety of different mouse models have been created referring to molecules of the mGluR1 signaling complex, including mGluR1, Gαq, PLC, PKCγ, ITPR1 and TRPC3 [4][5][6][7][8][9][10][11][12][13]. Some of these molecules are known to cause SCAs or other disorders relating to the cerebellum. For example, PKCγ mutants cause SCA14 [14][15][16] and PKCγ is downstream of mGluR1 signaling. SCA15 is caused by mutations of ITPR1 [17], another downstream molecule of mGluR1. In SCA1, mGluR1, ITPR1, PKCγ and Homer3 have been found to be downregulated on the transcriptional level [2][18][19] and for mGluR1 this reduction of expression has been confirmed on the protein level [20]. Disruption of mGluR1 has been reported in the mouse models of SCA3 [21] and SCA5 [22]. Mutations within GRM1 coding for mGluR1 are relatively rare. Recently, SCA44 has been reported, with heterozygous dominant mutations in the GRM1 gene showing typical phenotypes of SCA disease, but with different characteristics, possibly due to different functional changes in different mutants [23]. The function of an mGluR1 truncation mutation was tested by cellular experiments and this mutation resulted in a decreased receptor activity and decreased downstream target phosphorylation, suggesting that a loss of function of this mutation interferes with downstream signaling of mGluR1 [23]. These findings demonstrate an important role of mGluR1 signaling in Purkinje cells and show the relationship of altered mGluR1 signaling and SCA pathogenesis.
Evidence for enhanced mGluR1 signaling is also present in different types of SCA. For example, elevated calcium is reported in Purkinje cells of the SCA2 mouse model caused by ATXN2Q58 [24] and mutant ataxin2 can interact with IP3R [25]. TheOur previous study has reported increased IP3R1 expression (encoded by ITPR1, causative gene of SCA15 and SCA29) in an SCA14 mouse model [26]. The two other SCA44 causing mGluR1 missense mutations showed increased receptor activity compared to wild type mGluR1, suggesting that increased activity of mGluR1 leads to increased ligand sensitivity and ligand-independent activation, which is related to the fact that both missense mutations are closely located in the area of mGluR1 activation, resulting in excessive mGluR1 signaling by positive feedback with increased intracellular calcium levels [23]. The moonwalker (Mwk) mouse model with severe ataxia and abnormal Purkinje cell development, is caused by point mutations of TRPC3 (the causative gene of SCA41). This mutant TRPC3 can activate the cation channel, downstream of mGluR1 signaling [27][28]. These studies suggest that an increased activity of the mGluR1 pathway might also be associated with pathogenesis in some types of SCAs.
2. The Role of Recently Identified Dysregulated Molecules
Researchers have used the strategy of overlapping microarray data from different types of SCAs. Recently, these mouse models have been used to identify some key molecules which could contribute to uncover potentially shared molecular mechanisms related to the pathogenesis of SCAs [29][30][31][32].
2.1. RGS8
RGS8 is dysregulated in SCA1, SCA2, SCA7 and SCA14, indicating a role in pathogenesis of SCAs [29][30][31][33]. RGS8 has been reported to be strongly expressed in rat cerebellar Purkinje cells and it appears to be enriched in brainstem and nucleus accumbens [34][35][36][37]. RGS8 mRNA selectively interacts with ATXN2 and mutant ATXN2 reduced RGS8 expression in the SCA2 mouse model [31]. In a mouse model of SCA14 with increased PKCγ activity, RGS8 function has been studied in more detail. The increased RGS8 expression could partially counteract the negative effect of activated mGluR1 signaling during Purkinje cell development [30]. Since RGS8 is belonging to the R4 subfamily, its function is directly linked to Gq protein. RGS8 inhibits the M1 muscarinic acetylcholine receptor-Gq-mediated signaling in Xenopus oocytes [38] and has a strong inhibitory function for Gαq- and Gαi/o-dependent receptor activity [39]. However, RGS8 was also demonstrated to function via direct interaction with the relative receptor. RGS8 is able to interact with the third intracellular loop of melanin-concentrating hormone (MCH) receptor 1 (MCHR1) and inhibits the calcium mobilization induced by melanin-concentrating hormone [39]. Increased RGS8 expression through the inhibition of the MCHR1 signaling in the hippocampal CA1 region may be related to the antidepressant-like behavior of RGS8 transgenic mice [40]. Although the expression of RGS8 protein has been reported in the hippocampal CA1 region, RGS8 knockout mice have normal brain development and no major abnormalities in other organs [40][41]. RGS8 protein is also expressed in testis, but RGS8 knockout mice are viable and fertile [41]. Electroconvulsive seizures in rats caused an increase in RGS8 mRNA expression in the prefrontal cortex [42], suggesting a potential role for RGS8 in seizures.
2.2. INPP5A
INPP5A protein is identified as a common molecule dysregulated in SCA1, SCA2, SCA7, SCA14 and SCA17 [29][30][31][32]. The molecule INPP5A is an enzyme of the inositol polyphosphate 5 phosphatase family. In cellular signaling, INPP5A functions as an enzyme that inactivates IP3 to terminate downstream signaling [43][44][45]. The absence of Inpp5a protein leads to progressive degeneration of Purkinje cells. In SCA17 knock-in mice, reduced Inpp5a expression was reported, which was associated with increased IP3 levels. Importantly, overexpression of Inpp5a gene causes a reduction of IP3 levels in the cerebellum and rescues the symptoms of Purkinje cell degeneration in SCA17 mice [32]. Similar to SCA2, overexpression of Inpp5a alleviates Purkinje cell degeneration in SCA2 mice [46].
2.3. STK17B
Down-regulated mRNA of STK17B is found in SCA1, SCA7 and SCA41 mouse models. In a recent study about STK17B gene function in Purkinje cells, STK17B signaling has been identified as a downstream effector of PKCγ. Reduction of STK17B protein is confirmed specifically in the Purkinje cells from SCA14 mouse models [29][47][48]. STK17B, also known as DAP kinase-related apoptotic kinase 2 (DRAK2), is located on chromosome 2 (2q32.3) and was first isolated from human placenta and liver cDNA libraries [49]. It belongs to the serine/threonine kinase family of the death associated proteins (DAP). STK17B gene is related to STK17A gene, also known as DRAK1 gene, and both of them may constitute of a novel sub-family, which was originally thought to have the function of inducing apoptosis [49]. The STK17B protein structure includes an N-terminal autophosphorylation region and a C-terminal region with a nuclear localization signal. Another putative nuclear localization signal has been reported in the kinase domain [50]. STK17B has been relatively well studied in immunology as it is expressed in the immune system. The findings from the immune system may provide further insight for future studies on the function of STK17B in the brain. STK17B has been associated with calcium mobilization and homeostasis [50][51]. As the exact role of STK17B in brain is currently unclear, further studies would be helpful to better understand the function of STK17B in the nervous system in the future. STK17B protein has been reported to have a prominent expression in the brain, including the olfactory lobe, pituitary, superchiasmatic nuclei, ventricular zone and cerebellum [52]. Importantly, increased phosphorylation of STK17B protein causes negative effects on cerebellar Purkinje cell dendritic development. This negative effect can be partially rescued by a newly designed STK17B protein inhibitor Cpd16 [48]. These new findings give more insights for the treatment of SCAs. The new inhibitor could also be a potential drug for SCAs.
Taken together, these studies add new molecules associated with the alteration of a calcium channel and a compound to pharmaceutically manipulate mGluR1 signaling (Figure 1), which is thought to be an important factor in the pathology of SCAs.

Figure 1. The recently reported molecules RGS8, Inpp5a and Stk17b are involved in the mGluR1-PKCγ signaling pathway in the cerebellum. A new drug, Cpd16 has been shown to work as inhibitor to Stk17b, which can regulate the downstream mGluR1 signaling pathway.
3. Shared mGluR1-PKCγ Signaling Pathway in Spinocerebellar Ataxias
Since the different SCAs share the same cerebellar phenotypes, it is reasonable to assume that the underlying common pathogenic signaling could refer to the pathogenic features in the cerebellum, e.g., Purkinje cell degeneration. Through the review of recent studies of SCA mouse models, reswearchers summarize and find that mGluR1-PKCγ signaling is a common pathway that is dysregulated early in the onset of SCAs and is associated with Purkinje cell dendritic development.
Dysregulated expression of common molecules is not only related to Purkinje cell dendritic development, but to the pathology of disease. This indicates similar signaling events which occur in the early stage of disease. For the group I type, e.g., SCA1, SCA2, most of their SCA genes are responsible for transcription [53]. A change of mGluR1-PKCγ signaling should be an indirect response during the early state of the disease. For the group II type, the SCA-genes are directly pointing to shared signaling or relevant downstream signaling, such as mGluR1, PKCγ, ITPR1, and TRPC3 protein [6][7][9][10][11][13]. Elucidating these shared pathways will help researcherus to possibly modulate and monitor pathogenesis of different SCAs (Figure 2).
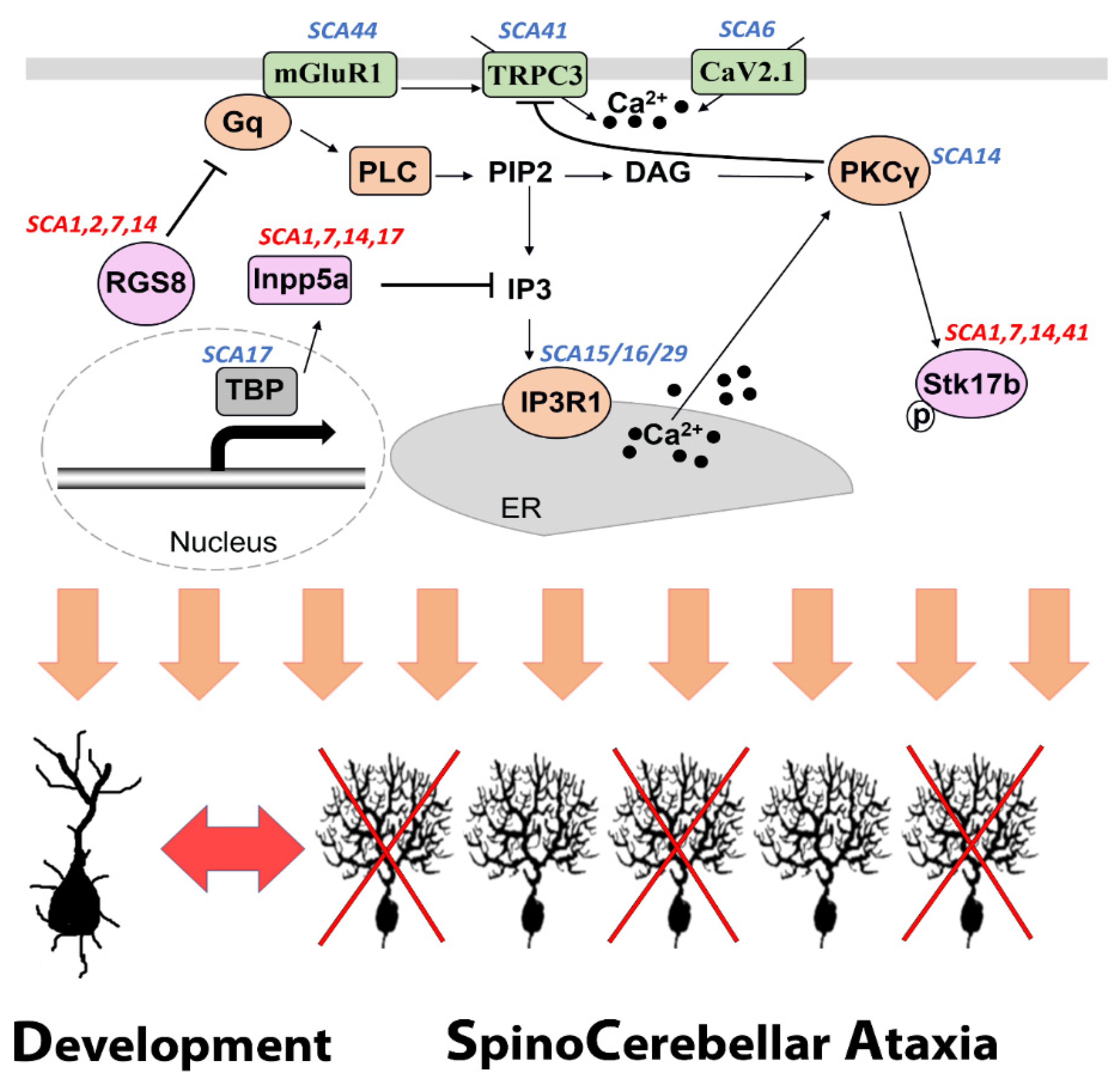
Figure 2. A shared mGluR1-PKCγ signaling pathway associated with SCA pathogenesis and Purkinje cell dendritic development. Several proteins which are components of the mGluR1-PKCγ signaling pathway are encoded by genes mutations of which are known to be causing SCA (labeled in blue). The common molecules RGS8, Inpp5a and Stk17b have been reported to be dysregulated in at least four different forms of SCA (labeled in red) and are directly involved in the mGluR1-PKCγ signaling pathway.
Although the alteration of mGluR1-PKCγ signaling is thought to affect dendritic development of Purkinje cells and to occur much earlier than at disease onset, dendritic development of Purkinje cells may not be the direct cause of the adult ataxia phenotype. The cause of ataxia is thought to be related to changes in the firing pattern of mature Purkinje cells. For example, deletion of the SCA13-associated gene KCNC3 affects the frequency of firing of Purkinje cells and increases the excitability of Purkinje cell dendrites [54]. Since mouse models of ataxia share the alteration of output signals in Purkinje cells, researchers have found that proper function of mGluR1 receptors and mGluR1 signaling are involved in the prevention of ataxia, which in turn suggests a link between the mGluR1 signaling pathway and ataxias. Further studies have shown that gene mutations related to mGluR1 or related signaling molecules cause the failure of climbing fiber maturation during the establishment of Purkinje cell innervation [55]. However, the suggested mGluR1 signaling pathway is certainly not able to explain the mechanism of all types of SCA. In a mouse model of SCA27, there was no significant change in the mGluR1 response, but the AMPA-mediated currents were impaired [56], suggesting that the mGluR1 pathway is not unique and that there are other potential signaling pathways that may cause SCAs.
Recently, an adult-stage RNA profiling was presented in a study using the SCA2 mouse model, with the expectation of finding signaling pathways important for Purkinje cell degeneration. Inpp5a and RGS8 have also been identified as dysregulated molecules in this adult mouse study. In addition, there are also many molecules associated with calcium signaling, such as Camk2a and Camk4, which have been described as downstream candidate factors of mGluR1 signaling in synaptic function [57][58]. Although these molecules have not been identified in the developmental phase and few studies have reported functions of these molecules for dendritic development of Purkinje cells, it is worthwhile to investigate them to gain a deeper understanding of SCA pathogenesis.
Serological testing using antibodies could help neurologists to diagnose cerebellar ataxias in clinical practice. Recent studies have shown that selective antibodies against molecules of the mGluR1 pathway are often present in patients with Autoimmune Cerebellar Ataxia. Importantly, many of these antigens are also associated with the pathogenesis of spinocerebellar ataxias [59][60][61]. This clinical evidence suggests that the mGluR1 signaling pathway may be a common pathophysiological mechanism not only in SCAs but also in other conditions with signs of cerebellar ataxia.
References
- Steinmayr, M.; André, E.; Conquet, F.; Rondi-Reig, L.; Delhaye-Bouchaud, N.; Auclair, N.; Daniel, H.; Crépel, F.; Mariani, J.; Sotelo, C.; et al. Staggerer Phenotype in Retinoid-Related Orphan Receptor α-Deficient Mice. Proc. Natl. Acad. Sci. USA 1998, 95, 3960–3965.
- Serra, H.G.; Duvick, L.; Zu, T.; Carlson, K.; Stevens, S.; Jorgensen, N.; Lysholm, A.; Burright, E.; Zoghbi, H.Y.; Clark, H.B.; et al. RORα-Mediated Purkinje Cell Development Determines Disease Severity in Adult SCA1 Mice. Cell 2006, 127, 697–708.
- Gold, D.A.; Baek, S.H.; Schork, N.J.; Rose, D.W.; Larsen, D.L.D.; Sachs, B.D.; Rosenfeld, M.G.; Hamilton, B.A. RORα Coordinates Reciprocal Signaling in Cerebellar Development through Sonic Hedgehog and Calcium-Dependent Pathways. Neuron 2003, 40, 1119–1131.
- van de Leemput, J.; Chandran, J.; Knight, M.A.; Holtzclaw, L.A.; Scholz, S.; Cookson, M.R.; Houlden, H.; Gwinn-Hardy, K.; Fung, H.C.; Lin, X.; et al. Deletion at ITPR1 Underlies Ataxia in Mice and Spinocerebellar Ataxia 15 in Humans. PLoS Genet. 2007, 3, 1076–1082.
- Aiba, A.; Chen, C.; Herrup, K.; Rosenmund, C.; Stevens, C.F.; Tonegawa, S. Reduced Hippocampal Long-Term Potentiation and Context-Specific Deficit in Associative Learning in MGluR1 Mutant Mice. Cell 1994, 79, 365–375.
- Conquet, F.; Bashir, Z.I.; Davies, C.H.; Daniel, H.; Ferraguti, F.; Bordi, F.; Franz-Bacon, K.; Reggiani, A.; Matarese, V.; Condé, F.; et al. Motor Deficit and Impairment of Synaptic Plasticity in Mice Lacking MGluR1. Nature 1994, 372, 237–243.
- Offermanns, S.; Toombs, C.F.; Hu, Y.H.; Simon, M.I. Defective Platelet Activation in Gα(q)-Deficient Mice. Nature 1997, 389, 183–186.
- Hartmann, J.; Blum, R.; Kovalchuk, Y.; Adelsberger, H.; Kuner, R.; Durand, G.M.; Miyata, M.; Kano, M.; Offermanns, S.; Konnerth, A. Distinct Roles of Gαq Gα11 and Purkinje Cell Signaling and Motor Behavior. J. Neurosci. 2004, 24, 5119–5130.
- Kano, M.; Hashimoto, K.; Watanabe, M.; Kurihara, H.; Offermanns, S.; Jiang, H.; Wu, Y.; Jun, K.; Shin, H.S.; Inoue, Y.; et al. Phospholipase Cβ4 Is Specifically Involved in Climbing Fiber Synapse Elimination in the Developing Cerebellum. Proc. Natl. Acad. Sci. USA 1998, 95, 15724–15729.
- Miyata, M.; Kim, H.T.; Hashimoto, K.; Lee, T.K.; Cho, S.Y.; Jiang, H.; Wu, Y.; Jun, K.; Wu, D.; Kano, M.; et al. Deficient Long-Term Synaptic Depression in the Rostral Cerebellum Correlated with Impaired Motor Learning in Phospholipase C Β4 Mutant Mice. Eur. J. Neurosci. 2001, 13, 1945–1954.
- Matsumoto, M.; Nakagawa, T.; Inoue, T.; Nagata, E.; Tanaka, K.; Takano, H.; Minowa, O.; Kuno, J.; Sakakibara, S.; Yamada, M.; et al. Ataxia and Epileptic Seizures in Mice Lacking Type 1 Inositol 1,4,5-Trisphosphate Receptor. Nature 1996, 379, 168–171.
- Hartmann, J.; Dragicevic, E.; Adelsberger, H.; Henning, H.A.; Sumser, M.; Abramowitz, J.; Blum, R.; Dietrich, A.; Freichel, M.; Flockerzi, V.; et al. TRPC3 Channels Are Required for Synaptic Transmission and Motor Coordination. Neuron 2008, 59, 392–398.
- Hartmann, J.; Henning, H.A.; Konnerth, A. MGluR1/TRPC3-Mediated Synaptic Transmission and Calcium Signaling in Mammalian Central Neurons. Cold Spring Harb. Perspect. Biol. 2011, 3, a006726.
- Ji, J.; Hassler, M.L.; Shimobayashi, E.; Paka, N.; Streit, R.; Kapfhammer, J.P. Increased Protein Kinase C Gamma Activity Induces Purkinje Cell Pathology in a Mouse Model of Spinocerebellar Ataxia 14. Neurobiol. Dis. 2014, 70, 1–11.
- Trzesniewski, J.; Altmann, S.; Jäger, L.; Kapfhammer, J.P. Reduced Purkinje Cell Size Is Compatible with near Normal Morphology and Function of the Cerebellar Cortex in a Mouse Model of Spinocerebellar Ataxia. Exp. Neurol. 2019, 311, 205–212.
- Shimobayashi, E.; Kapfhammer, J.P. Increased Biological Activity of Protein Kinase C Gamma Is Not Required in Spinocerebellar Ataxia 14. Mol. Brain 2017, 10, 34.
- Hara, K.; Shiga, A.; Nozaki, H.; Mitsui, J.; Takahashi, Y.; Ishiguro, H.; Yomono, H.; Kurisaki, H.; Goto, J.; Ikeuchi, T.; et al. Total Deletion and a Missense Mutation of ITPR1 in Japanese SCA15 Families. Neurology 2008, 71, 547–551.
- Lin, X.; Antalffy, B.; Kang, D.; Orr, H.T.; Zoghbi, H.Y. Polyglutamine Expansion Down-Regulates Specific Neuronal Genes before Pathologic Changes in SCA1. Nat. Neurosci. 2000, 3, 157–163.
- Serra, H.G.; Byam, C.E.; Lande, J.D.; Tousey, S.K.; Zoghbi, H.Y.; Orr, H.T. Gene Profiling Links SCA1 Pathophysiology to Glutamate Signaling in Purkinje Cells of Transgenic Mice. Hum. Mol. Genet. 2004, 13, 2535–2543.
- Zu, T.; Duvick, L.A.; Kaytor, M.D.; Berlinger, M.S.; Zoghbi, H.Y.; Clark, H.B.; Orr, H.T. Recovery from Polyglutamine-Induced Neurodegeneration in Conditional SCA1 Transgenic Mice. J. Neurosci. 2004, 24, 8853–8861.
- Konno, A.; Shuvaev, A.N.; Miyake, N.; Miyake, K.; Iizuka, A.; Matsuura, S.; Huda, F.; Nakamura, K.; Yanagi, S.; Shimada, T.; et al. Mutant Ataxin-3 with an Abnormally Expanded Polyglutamine Chain Disrupts Dendritic Development and Metabotropic Glutamate Receptor Signaling in Mouse Cerebellar Purkinje Cells. Cerebellum 2014, 13, 29–41.
- Armbrust, K.R.; Wang, X.; Hathorn, T.J.; Cramer, S.W.; Chen, G.; Zu, T.; Kangas, T.; Zink, A.N.; Öz, G.; Ebner, T.J.; et al. Mutant β-III Spectrin Causes MGluR1α Mislocalization and Functional Deficits in a Mouse Model of Spinocerebellar Ataxia Type 5. J. Neurosci. 2014, 34, 9891–9904.
- Watson, L.M.; Bamber, E.; Schnekenberg, R.P.; Williams, J.; Bettencourt, C.; Lickiss, J.; Fawcett, K.; Clokie, S.; Wallis, Y.; Clouston, P.; et al. Dominant Mutations in GRM1 Cause Spinocerebellar Ataxia Type 44. Am. J. Hum. Genet. 2017, 101, 451–458.
- Liu, J.; Tang, T.S.; Tu, H.; Nelson, O.; Herndon, E.; Huynh, D.P.; Pulst, S.M.; Bezprozvanny, I. Deranged Calcium Signaling and Neurodegeneration in Spinocerebellar Ataxia Type 2. J. Neurosci. 2009, 29, 9148–9162.
- Kasumu, A.W.; Hougaard, C.; Rode, F.; Jacobsen, T.A.; Sabatier, J.M.; Eriksen, B.L.; Strobæk, D.; Liang, X.; Egorova, P.; Vorontsova, D.; et al. Selective Positive Modulator of Calcium-Activated Potassium Channels Exerts Beneficial Effects in a Mouse Model of Spinocerebellar Ataxia Type 2. Chem. Biol. 2012, 19, 1340–1353.
- Shimobayashi, E.; Wagner, W.; Kapfhammer, J.P. Carbonic Anhydrase 8 Expression in Purkinje Cells Is Controlled by PKCγ Activity and Regulates Purkinje Cell Dendritic Growth. Mol. Neurobiol. 2016, 53, 5149–5160.
- Becker, E.B.E.; Oliver, P.L.; Glitsch, M.D.; Banks, G.T.; Achillic, F.; Hardy, A.; Nolan, P.M.; Fisher, E.M.C.; Davies, K.E. A Point Mutation in TRPC3 Causes Abnormal Purkinje Cell Development and Cerebellar Ataxia in Moonwalker Mice. Proc. Natl. Acad. Sci. USA 2009, 106, 6706–6711.
- Sekerková, G.; Kim, J.A.; Nigro, M.J.; Becker, E.B.E.; Hartmann, J.; Birnbaumer, L.; Mugnaini, E.; Martina, M. Early Onset of Ataxia in Moonwalker Mice Is Accompanied by Complete Ablation of Type II Unipolar Brush Cells and Purkinje Cell Dysfunction. J. Neurosci. 2013, 33, 19689–19694.
- Gatchel, J.R.; Watase, K.; Thaller, C.; Carson, J.P.; Jafar-Nejad, P.; Shaw, C.; Zu, T.; Orr, H.T.; Zoghbi, H.Y. The Insulin-like Growth Factor Pathway Is Altered in Spinocerebellar Ataxia Type 1 and Type 7. Proc. Natl. Acad. Sci. USA 2008, 105, 1291–1296.
- Wu, Q.-W.; Kapfhammer, J.P. Modulation of Increased MGluR1 Signaling by RGS8 Protects Purkinje Cells From Dendritic Reduction and Could Be a Common Mechanism in Diverse Forms of Spinocerebellar Ataxia. Front. Cell Dev. Biol. 2021, 8, 1912.
- Dansithong, W.; Paul, S.; Figueroa, K.P.; Rinehart, M.D.; Wiest, S.; Pflieger, L.T.; Scoles, D.R.; Pulst, S.M. Ataxin-2 Regulates RGS8 Translation in a New BAC-SCA2 Transgenic Mouse Model. PLoS Genet. 2015, 11, e1005182.
- Liu, Q.; Huang, S.; Yin, P.; Yang, S.; Zhang, J.; Jing, L.; Cheng, S.; Tang, B.; Li, X.J.; Pan, Y.; et al. Cerebellum-Enriched Protein INPP5A Contributes to Selective Neuropathology in Mouse Model of Spinocerebellar Ataxias Type 17. Nat. Commun. 2020, 11, 1101.
- Wu, Q.-W.; Kapfhammer, J.P. CRISPR-Cas13-Mediated Knockdown of Regulator of G-Protein Signaling 8 (RGS8) Does Not Affect Purkinje Cell Dendritic Development. Front. Cell Dev. Biol. 2022, 10, 854273.
- Larminie, C.; Murdock, P.; Walhin, J.P.; Duckworth, M.; Blumer, K.J.; Scheideler, M.A.; Garnier, M. Selective Expression of Regulators of G-Protein Signaling (RGS) in the Human Central Nervous System. Mol. Brain Res. 2004, 122, 24–34.
- Gold, S.J.; Ni, Y.G.; Dohlman, H.G.; Nestler, E.J. Regulators of G-Protein Signaling (RGS) Proteins: Region-Specific Expression of Nine Subtypes in Rat Brain. J. Neurosci. 1997, 17, 8024–8037.
- Saitoh, O.; Odagiri, M. RGS8 Expression in Developing Cerebellar Purkinje Cells. Biochem. Biophys. Res. Commun. 2003, 309, 836–842.
- Saitoh, O.; Masuho, I.; Itoh, M.; Abe, H.; Komori, K.; Odagiri, M. Distribution of Regulator of G Protein Signaling 8 (RGS8) Protein in the Cerebellum. Cerebellum 2003, 2, 154–160.
- Itoh, M.; Nagatomo, K.; Kubo, Y.; Saitoh, O. Alternative Splicing of RGS8 Gene Changes the Binding Property to the M1 Muscarinic Receptor to Confer Receptor Type-Specific Gq Regulation. J. Neurochem. 2006, 99, 1505–1516.
- Miyamoto-Matsubara, M.; Saitoh, O.; Maruyama, K.; Aizaki, Y.; Saito, Y. Regulation of Melanin-Concentrating Hormone Receptor 1 Signaling by RGS8 with the Receptor Third Intracellular Loop. Cell. Signal. 2008, 20, 2084–2094.
- Kobayashi, Y.; Takemoto, R.; Yamato, S.; Okada, T.; Iijima, M.; Uematsu, Y.; Chaki, S.; Saito, Y. Depression-Resistant Phenotype in Mice Overexpressing Regulator of G Protein Signaling 8 (RGS8). Neuroscience 2018, 383, 160–169.
- Kuwata, H.; Nakao, K.; Harada, T.; Matsuda, I.; Aiba, A. Generation of RGS8 Null Mutant Mice by Cre/LoxP System. Kobe J. Med. Sci. 2007, 53, 275–281.
- Gold, S.J.; Heifets, B.D.; Pudiak, C.M.; Potts, B.W.; Nestler, E.J. Regulation of Regulators of G Protein Signaling MRNA Expression in Rat Brain by Acute and Chronic Electroconvulsive Seizures. J. Neurochem. 2002, 82, 828–838.
- Berridge, M.J. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol. Rev. 2016, 96, 1261–1296.
- Furuichi, T.; Mikoshiba, K. Inositol 1,4,5-Trisphosphate Receptor-Mediated Ca2+ Signaling in the Brain. J. Neurochem. 1995, 64, 953–960.
- Mitchell, C.A.; Gurung, R.; Kong, A.M.; Dyson, J.M.; Tan, A.; Ooms, L.M. Inositol Polyphosphate 5-Phosphatases: Lipid Phosphatases With Flair. IUBMB Life 2002, 53, 25–36.
- Kasumu, A.W.; Liang, X.; Egorova, P.; Vorontsova, D.; Bezprozvanny, I. Chronic Suppression of Inositol 1,4,5-Triphosphate Receptor-Mediated Calcium Signaling in Cerebellar Purkinje Cells Alleviates Pathological Phenotype in Spinocerebellar Ataxia 2 Mice. J. Neurosci. 2012, 32, 12786.
- Dulneva, A.; Lee, S.; Oliver, P.L.; di Gleria, K.; Kessler, B.M.; Davies, K.E.; Becker, E.B.E. The Mutant Moonwalker TRPC3 Channel Links Calcium Signaling to Lipid Metabolism in the Developing Cerebellum. Hum. Mol. Genet. 2015, 24, 4114–4125.
- Wu, Q.-W.; Kapfhammer, J.P. Serine/Threonine Kinase 17b (STK17B) Signalling Regulates Purkinje Cell Dendritic Development and Is Altered in Multiple Spinocerebellar Ataxias. Eur. J. Neurosci. 2021, 54, 6673–6684.
- Sanjo, H.; Kawait, T.; Akira, S. DRAKs, Novel Serine/Threonine Kinases Related to Death-Associated Protein Kinase That Trigger Apoptosis. J. Biol. Chem. 1998, 273, 29066–29071.
- Friedrich, M.L.; Wen, B.G.; Bain, G.; Kee, B.L.; Katayama, C.; Murre, C.; Hedrick, S.M.; Walsh, C.M. DRAK2, a Lymphoid-Enriched DAP Kinase, Regulates the TCR Activation Threshold during Thymocyte Selection. Int. Immunol. 2005, 17, 1379–1390.
- McGargill, M.A.; Wen, B.G.; Walsh, C.M.; Hedrick, S.M. A Deficiency in Drak2 Results in a T Cell Hypersensitivity and an Unexpected Resistance to Autoimmunity. Immunity 2004, 21, 781–791.
- Mao, J.; Qiao, X.; Luo, H.; Wu, J. Transgenic Drak2 Overexpression in Mice Leads to Increased T Cell Apoptosis and Compromised Memory T Cell Development. J. Biol. Chem. 2006, 281, 12587–12595.
- Klockgether, T.; Mariotti, C.; Paulson, H.L. Spinocerebellar Ataxia. Nat. Rev. Dis. Primers 2019, 5, 24.
- Zagha, E.; Manita, S.; Ross, W.N.; Rudy, B. Dendritic Kv3.3 Potassium Channels in Cerebellar Purkinje Cells Regulate Generation and Spatial Dynamics of Dendritic Ca2+ Spikes. J. Neurophysiol. 2010, 103, 3516–3525.
- Hoxha, E.; Balbo, I.; Miniaci, M.C.; Tempia, F. Purkinje Cell Signaling Deficits in Animal Models of Ataxia. Front. Synaptic Neurosci. 2018, 10, 6.
- Tempia, F.; Hoxha, E.; Negro, G.; Alshammari, M.A.; Alshammari, T.K.; Panova-Elektronova, N.; Laezza, F. Parallel Fiber to Purkinje Cell Synaptic Impairment in a Mouse Model of Spinocerebellar Ataxia Type 27. Front. Cell. Neurosci. 2015, 9, 205.
- Ribar, T.J.; Rodriguiz, R.M.; Khiroug, L.; Wetsel, W.C.; Augustine, G.J.; Means, A.R. Cerebellar Defects in Ca2+/Calmodulin Kinase IV-Deficient Mice. J. Neurosci. 2000, 20, RC107.
- Arsović, A.; Halbach, M.V.; Canet-Pons, J.; Esen-Sehir, D.; Döring, C.; Freudenberg, F.; Czechowska, N.; Seidel, K.; Baader, S.L.; Gispert, S.; et al. Mouse Ataxin-2 Expansion Downregulates CamKII and Other Calcium Signaling Factors, Impairing Granule—Purkinje Neuron Synaptic Strength. Int. J. Mol. Sci. 2020, 21, 6673.
- Jarius, S.; Wildemann, B. “Medusa-Head Ataxia”: The Expanding Spectrum of Purkinje Cell Antibodies in Autoimmune Cerebellar Ataxia. Part 1: Anti-MGluR1, Anti-Homer-3, Anti-Sj/ITPR1 and Anti-CARP VIII. J. Neuroinflamm. 2015, 12, 168.
- Mitoma, H.; Honnorat, J.; Yamaguchi, K.; Manto, M. Cerebellar Long-Term Depression and Auto-Immune Target of Auto-Antibodies: The Concept of LTDpathies. Mol. Biomed. 2021, 2, 2.
- Garza, M.; Piquet, A.L. Update in Autoimmune Movement Disorders: Newly Described Antigen Targets in Autoimmune and Paraneoplastic Cerebellar Ataxia. Front. Neurol. 2021, 12, 683048.
More