Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Stephanie Duguez and Version 3 by Beatrix Zheng.
Amyotrophic Lateral Sclerosis (ALS), Spinal Bulbar Muscular Atrophy (SBMA), and Spinal Muscular Atrophy (SMA) are motor neuron diseases (MNDs) characterised by progressive motor neuron degeneration, weakness and muscular atrophy. Lipid dysregulation is well recognised in each of these conditions and occurs prior to neurodegeneration. Several lipid markers have been shown to predict prognosis in ALS. Sphingolipids are complex lipids enriched in the central nervous system and are integral to key cellular functions including membrane stability and signalling pathways, as well as being mediators of neuroinflammation and neurodegeneration.
- sphingomyelin
- ceramide
- Amyotrophic Lateral Sclerosis
- sphingolipid
- motor neuron disease
1. Sphingolipid Synthesis
Sphingolipids (SLs) are a diverse class of lipids with eighteen carbon amino-alcohol backbones, which are synthesized in the ER from non-sphingolipid precursors [1][39]. They play significant roles in membrane structure and have many bioactive metabolites, which regulate cellular function [1][2][39,40]. The basic structure of SLs is ceramide. Ceramide consists of a sphingoid long-chain base and a fatty acid acyl chain connected to an amine bond [3][41]. The most common mammalian long-chain base is sphingosine (d18:1), an 18 Carbon chain with a trans double bond at positions 4–5 [4][42]. The structure of sphingosine, ceramide and SM are shown in Figure 1.
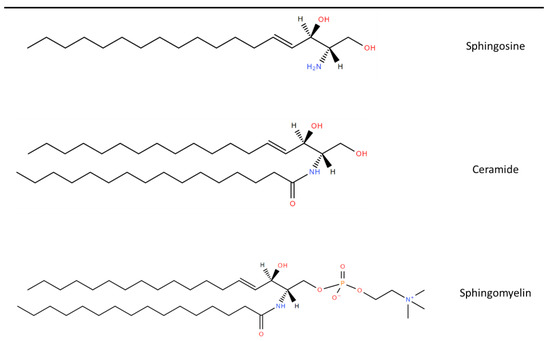
Figure 1. Chemical structure of common Sphingolipids. Sphingosine is the most common long-chain base. A fatty acid acyl chain is connected to the C2 amide group to form Ceramide and then Sphingomyelin is formed by the subsequent addition of a phosphocholine head group. Lipid structures created using LIPID MAPS® tools [5][43].
2. Role of Sphingolipids in MNDs
Given the roles of SLs in many vital biological processes and their high abundance in the central nervous system as major components of oligodendrocytes and myelin sheaths, SL metabolism is thought to be a key pathway in neurodegeneration and neuroinflammation [6][83]. Alteration in SL metabolism has been linked to multiple neurodegenerative diseases such as Alzheimer’s Disease and Parkinson’s Disease, as well as neuroinflammatory conditions such as Multiple Sclerosis. These are discussed in detail in other reviews [7][8][9][84,85,86].
Increased levels of SM and ceramide have been found in spinal cord tissue of patients with Amyotrophic Lateral Sclerosis (ALS) S and SOD1 mice [10][16]. A study in the wobbler mouse, which is a model of motor neuron degeneration, identified the mis-sorting of lysosomal SL degradation enzymes with a resultant increase in SL intermediates [11][87]. Lipid dysregulation in ALS can occur decades before classical symptoms, and lipid biomarkers can be used to identify individuals at risk of developing ALS [12][13][24,88]. In keeping with this, increased levels of SLs were identified in spinal cords of ALS mice prior to the onset of clinical signs, and SM was demonstrated to mediate motor neuron death via oxidative stress [10][16]. A transcriptomic meta-analysis study on spinal cord tissue from SOD1 mice found that cholesterol, ceramides and eicosanoid pathways were altered early in the disease course [14][89]. This has also been shown in human studies, with SL alteration identified in plasma samples of patients who subsequently developed ALS [15][90]. Importantly, these studies suggest that alterations in SL occur before motor neuron degeneration and are therefore an upstream process in ALS pathophysiology.
Over 20 risk genes in ALS are involved in lipid raft homeostasis and ceramide metabolic pathways [16][58]. Mutations or abnormal DNA methylation have been found in genes encoding for enzymes necessary for SL synthesis in patients with ALS and Spinal Muscular Atrophy (SMA)A, as well as bovine SMA. These are shown in Table 1. In addition, mutations in ASAH1, which result in dysfunctional acid ceramidase, cause a non-5q form of SMA associated with progressive myoclonic epilepsy [17][91]. Mutations in the SPTLC1 gene are associated with juvenile ALS and Hereditary Sensory and Autonomic Neuropathy type 1 (HSAN1) [18][19][92,93]. This gene encodes for a subunit of SPT, the enzyme required for the first step of SL synthesis. C-terminal SPTLC1 variants cause the formation of atypical deoxysphingolipids and result in HSAN1 [20][94]. The ALS-causing variants map to a transmembrane domain, which interacts with negative regulators of SPT activity and results in unregulated SPT and excess SL synthesis [21][95]. Epigenomic studies have also shown abnormal DNA methylation in SGMS2, which encodes for SMS2, the enzyme for converting ceramide to SM [22][96]. The CAV1 gene, which encodes for calveolin 1, has also recently been identified as a risk modifying gene in ALS. Calveolin 1 is found in lipid rafts and ALS variants in CAV1 were shown to disrupt lipid raft formation in patient-derived lymphoblastoid cells [23][97].
Table 1.
Abnormalities of Sphingolipid metabolism in Motor Neuron Diseases and sphingolipidoses.
| Condition | Gene | Affected Enzyme/Protein | Effect on Sphingolipids | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sphingolipid synthesis | ||||||||||
| Juvenile ALS [18][92] HSAN1 [19][93] |
SPTLC1 | SPT | Atypical deoxysphingolipids, cannot be converted into complex SLs or degraded | |||||||
| ↓: TG (16:1/18:1/18:2) | Bovine SMA [24][106] | FVT1 | ||||||||
| Lawton et al. 2012 [46][ | KSR | 126] | Reduced ceramide synthesis from de novo pathway | |||||||
| 161 ALS | 117 Controls |
Plasma | GC/MS and UPLC-MS/MS | 335 lipids, proteins and carbohydrates | ↑: LPC (16:1) and SM (18:0) | Not evaluated | ALS type 8 [25][107] Late onset SMA [26 | |||
| Cutler et al. 2002 [10] | ][ | [16 | 108] | ] | VAPB | VAPB with effect on CERT and FAPP2 | Impaired transfer of ceramide and glucosylceramide from ER to golgi apparatus | |||
| 9 ALS | 3 Control |
Spinal cord | ES/MS/MS | Sphingolipids, Phospholipids, Cholesterol Esters, and Lipid Peroxides |
↑: Cer (C16:0), Cer (C24:0), SM (C16:0), CE (C16:0) and CE (C18:0) | ALS [22][96] | SGMS2 | SMS2 | Affects sphingomyelin synthesis | |
| Sphingolipid degradation | ||||||||||
| Not evaluated | ||||||||||
| Goutman et al. 2020 [47] | SMA-PME [17][91] Farber’s disease [27][109] |
ASAH1 | Acid ceramidase | Ceramide accumulation | ||||||
| [127] | 125 ALS 71 Controls |
Plasma | UPLC-MS/MS | 899 metabolites | ↑: 8 Cers, 28 DAGs, 5 HEXC, 24 SMs, | Not evaluated | ||||
| ↓: 5 DAGs, 5 SMs | ||||||||||
| Goutman et al. 2022 [48][128] | Above cohort of 125 ALS and 71 controls with 2nd cohort 225 ALS, 104 controls | Plasma | UPLC-MS/MS | 640 metabolites | SM most significant sub-pathway LCFA, acyl intermediates and Cers also raised |
SM (d18:1/24:0), SM (d18:1/20:0, d16:1/22:0), SM (d18:1/14:0, d16:1/16:0) and lignoceroylcarnitine (C24) correlated with ALSFRS-R | GM1 gangliodosis [28][110] | GLB1 | β-Galactosidase | GM1 ganglioside accumulation |
| Bjornevik et al. 2019 [15][90] | 275 ALS | GM2 gangliodoses [28][110]
|
HEXA HEXB |
|
GM2 ganglioside accumulation GM2 ganglioside, glycolipid GA2 and globoside accumulation |
|||||
| Fabry’s Disease [29][111] | GLA | α-Galactosidase A | Globotriaosylceramide accumulation | |||||||
| Metachromatic Leukodystrophy [30][112] | ARSA | Arylsulphatase A | Sulfatides accumulation | |||||||
Niemann-Pick Disease [31][113]
|
SMPD1 NPC1/NPC2 |
Sphingomyelinase | Sphingomyelin accumulation | |||||||
| Gaucher’s Disease [32][114] | GBA | Glucocerebrosidase | Glucosylceramide accumulation | |||||||
| Krabbe’s Disease [33][115] | GALC | Galactosylceramidase | Galactosylceramide accumulation | |||||||
HSAN1—hereditary sensory and autonomic neuropathy type 1, SPT—Serine palmitoyltransferase, VAPB—Vesicle associated membrane protein B, CERT—ceramide transfer protein, FAPP2—four phosphate adapter protein 2, SMS2—sphingomyelin synthase 2, SMA-PME—spinal muscular atrophy and progressive myoclonic epilepsy.
Another mechanism of how SLs can affect MNDs is through intercellular communication. Neutral SMase2 affects EV secretion. This has been demonstrated by studies showing that stimulation of SMase2 with TNF alpha increases EV secretion and inhibiting it with 1 PDDC reduces EV secretion [34][35][82,98]. EVs are being increasingly investigated in ALS as mediators of intercellular transfer of neurotoxic proteins such as TDP 43, FUS and SOD1 [36][37][99,100]. EVs secreted by muscle cells from ALS patients have been shown to be toxic to motor neurons [38][101].
Further insight into the importance of SL metabolism in neurodegenerative diseases is evident from lysosomal storage disorders. These are a group of over 40 conditions with a combined prevalence of 1 in 7000–8000 live births [39][102]. These diseases all are the result of impaired lysosomal degradation of various metabolites and the consequent effects on cellular function [40][103]. Several involve the degradation of SLs and are termed sphingolipidoses. These are a group of autosomal recessive or X-linked conditions with defects in enzymes required for the catabolism of SLs [41][104]. The cellular impact of the conditions depends on the concentration of the relevant SL and the degree of enzymatic deficiency. The sphingolipidoses and their enzymatic defects and effects on SLs are shown in Table 1. They each have a broad and unique clinical phenotype. However, given that SLs are enriched in the nervous system, these conditions often have the predominant feature of severe progressive neurodegeneration [41][42][104,105].
3. Lipidomic Studies in MNDs
The lipid profiles in MNDs have been mainly assessed via metabolomic analysis. Table 2 lists all of the published metabolomic studies that have included lipidomic analysis to date, detailing a range of different SM and ceramides identified. This may in part be explained by the different samples studied and the differing mass spectrometry methodologies for quantifying metabolites. Two studies were performed using spinal cord tissue, nine using plasma, two using serum, and two using CSF samples. Of the 12 studies comparing ALS to controls, all identified changes in SM concentrations, with SM species being increased in 11 studies and decreased in the other. Six studies identified increases in ceramide species, with decreases in some ceramides reported in one of these. A study of only of ALS patients found that multiple SMs were able to predict markers of disease progression such as the ALSFRS-R, manual muscle testing and respiratory function [43][116]. Another metabolomic study in 28 patients with ALS and 30 controls reported that out of 317 metabolites, 50 were increased and 70 decreased in ALS, although the individual metabolites were not listed [44][117].
Table 2.
Metabolomic studies in patients with ALS showing the changes in lipid metabolites.
| Study | Patients | Sample Type | Quantification Platform | Metabolites Evaluated | Lipid Changes in MND | Prognostic Use |
|---|---|---|---|---|---|---|
| Blasco et al. 2017 [45][125] | 40 ALS 45 Controls |
CSF | HRMS | 122 lipids | ↑: PC (36:4p), PC (36:4e), SM (d43:2), SM (d34:0) | Higher SM (d43:2) and lower TG (16:0/16:0/18:1) and TG (18:0/16:0/18:1) had slower progression |
| 549 Controls | ||||||
| Plasma | ||||||
| LC/MS | ||||||
| 404 metabolites | ↑: SM (C18:2), PC (C40:7), PC (C38:4), CE (C22:4) | Not evaluated | ||||
| ↓: 12 TAGs, DAG (C36:1), DAG (C36:2), PC (C36:2), 21-deoxycortisol, butyrobetaine |
||||||
| Lawton et al. 2014 [49][122] | 172 ALS 73 neurological mimics 50 Controls |
plasma | GC/MS and UPLC-MS/MS | 367 metabolites | ↑: SM (d18:1/16:0), 5 FAs, 3-dehydrocarnitine, 1,2-propanediol, Chol, 1-stearoyl-GPI |
1,2-propanediol correlated with ALSFRS-R |
| Chang et al. 2021 [50][129] | 36 ALS 36 Controls |
plasma | LC–MS/MS | 185 metabolites | ↑: SM (C24:1), SM (C20:2), PC (C44:5), PC (C34:2) | 14 PCs and (OH) SM(C22:1) correlated with ALSFRS-R |
| ↓: (OH) SM(C22:1) (OH) SM(C24:1) 29 other PCs |
||||||
| Fernandez-Eulate et al. 2020 [51][130] | 20 ALS 20 Controls |
Serum | UPLC-MS | 416 lipids | ↑: SM (39:1), SM (33:1), PE (P-20:1/0:0), PE (O-16:0/0:0), 5 PCs, androsterone, etiocholanolone and 2 FAs |
Not evaluated |
| Blasco et al. 2018 [43][116] | 74 ALS | Plasma | HPLC-MS/MS | 188 metabolites | Not evaluated—no control participants | SM (C22:3) and SM (C34:1) correlated with disease progression, SM (24:1), SM (C16:1) and (OH) SM (C22:2) correlated with SVC |
| Dodge et al. 2015 [52][131] | 6 ALS 6 Control |
Spinal cord | LC-MS/MS | Cer, SM and GSLs | ↑: Cer (C18:0), Cer (C24:1), (OH) Cer (C24:0), Cerebroside (C18:0 and C24:1), GlcCer (C18:0 and C24:1), LacCer (18:0), GL3 (C22:1), GM3 (C23:0), GM1 (C18:0) AND SM (C18:0) | Not evaluated |
| Sol et al. 2021 [53][132] | 23 ALS 10 Controls |
CSF Plasma |
LC-MS/MS | 1018 lipids in plasma and 843 in CSF | ↑: 3 Fas, 2 DAGs, 13 TGs, 17 GPLs, 3 Cer, 1 SM | Fast vs. slow progressors had increased- 1 FA, 4 GLs, 4 GPLs, 2 Cer, 1 GM3, and decreased- 46 GLs, 36 GPLs, 2 Cer, 8 SM, 5 CE |
| ↓: 2 DAGs, 4 GPLs, 3 Cer, 3 GLs | ||||||
| Area-Gomez et al. 2021 [54][133] | 40 ALS 28 PLS 28 Control |
Serum/Plasma | LC/MS | 532 lipids | ↑: Cer, LacCer, CE | SM declined and Cer increased at follow up |
| ↓: SM, PC, PS |
HRMS—high-resolution mass spectrometry, GC/MS—gas chromatography/mass spectrometry, LC/MS—liquid chromatography/mass spectrometry, LC–MS/MS—liquid chromatography/tandem mass spectrometry, UPLC-MS/MS—ultra-high-performance liquid chromatography/tandem mass spectrometry, ES/MS/MS—electrospray ionization tandem mass spectrometry, CSF—cerebrospinal fluid, ALSFRS-R—Revised ALS Functional Rating Scale, SVC—slow vital capacity, SM—sphingomyelin, TG—triglyceride, LPC—palmitoleoyl-glycerophosphocholine, Cer—ceramide, CE—cholesterol ester, DAG-Diacylglycerol, HEXC—hexosylceramide, LCFA—long chain fatty acid, TAG—Triacylglycerol, PC—phosphatidylcholine, FA—fatty acids, GPI—glycophosphatidylinositol, (OH)SM—hydroxysphingomyelin, PE—phosphatidylethanolamines, PS—phosphatidylserines, GPL—glycerophospholipids, GL—glycerolipid.
One study identified four lipids, including SM C18:2, which were elevated several years before symptom onset [15][90]. This is of particular relevance with the progress in developing genotype-specific treatments, such as antisense oligonucleotides (ASOs) for patients with SOD1 and C9orf72 mutations and the need for biomarkers to guide the optimal timing for commencing treatment [55][56][57][118,119,120]. The ATLAS trial is currently evaluating Tofersen, an ASO for SOD1, in presymptomatic patients who develop raised neurofilament light chain levels, a marker of neuronal damage that becomes elevated 6–12 months prior to symptoms [58][121]. Given that lipids including SM and ceramide are altered early in the disease course [10][13][16,88], they could be of use in identifying presymptomatic patients for potential treatments. In addition, Blasco et al. have shown how SL biomarkers could be incorporated into pharmaco-metabolomic studies [43][116]. Baseline and follow-up SL profiles could be used to (1) further validate their use as prognostic markers compared to common clinical measurements of disease progression (such as lung function and ALSFRS-R) and (2) determine if treatments lead to alterations in metabolite levels.
There are little data on the lipidomic profile of other MNDs. There have been no lipidomic studies in Spinal Bulbar Muscular Atrophy (SBMA)MA. SBMA and SMA patients were included as neurological mimics in one study but were combined as part of a group containing other conditions such as cervical myelopathy and multiple sclerosis [49][122]. In a metabolomic study of patients with SMA, H-nuclear magnetic resonance-based metabolic profiling demonstrated diagnostic and prognostic utility, but individual metabolites were not listed [59][123]. Another metabolomic study in 108 patients with SMA showed 200 metabolites correlating with the modified Hammersmith functional motor scale, including 12 lipids. Only 1 lipid (SM (C24:1)) was among the top 20 metabolites identified [60][124].