The sideroblastic anemias are a heterogeneous group of inherited and acquired disorders characterized by anemia and the presence of ring sideroblasts in the bone marrow. Ring sideroblasts are abnormal erythroblasts with iron-loaded mitochondria that are visualized by Prussian blue staining as a perinuclear ring of green-blue granules. The mechanisms that lead to the ring sideroblast formation are heterogeneous, but in all of them, there is an abnormal deposition of iron in the mitochondria of erythroblasts. Congenital sideroblastic anemias include nonsyndromic and syndromic disorders. Acquired sideroblastic anemias include conditions that range from clonal disorders (myeloid neoplasms as myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms with ring sideroblasts) to toxic or metabolic reversible sideroblastic anemia. In the last 30 years, dDue to the advances in genomic techniques, a deep knowledge of the pathophysiological mechanisms has been accomplished and the bases for possible targeted treatments have been established. The distinction between the different forms of sideroblastic anemia is based on the study of the characteristics of the anemia, age of diagnosis, clinical manifestations, and the performance of laboratory analysis involving genetic testing in many cases.
- sideroblastic anemia
- ring sideroblast
- MDS
- MDS/MPN-RS-T
- SF3B1
1. Introduction
2. Historical Context
In 1942, Hans Grüneberg demonstrated, using the Prussian blue staining, the presence of free iron in the cytoplasm of some erythroblasts (sideroblasts) and in some mature erythrocytes (siderocytes) [2]. In 1945, Cooley [3] described a patient with sex-linked anemia, which probably corresponded to a case of the nonsyndromic form of X-linked SA (XLSA), since later Cotter et al. identified mutations in ALAS2 and ringed sideroblasts in the same family [4]. In 1956, Björkman described a series of four patients with chronic refractory anemia with numerous abnormal bone marrow sideroblasts, one of which developed leukemia. This is probably the first description of myelodysplastic syndromes with ring sideroblasts [5]. Sideroblastic anemias were recognized as a specific subtype of anemia in the 1960s [6]. Within the last 30 years, with the important advances in molecular biology, the genetic origin of more than two-thirds of congenital sideroblastic anemias cases, and a great proportion of cases of acquired clonal disease have been clarified.3. Ring Sideroblast Definition
Prussian blue staining (Perls’ reaction) is an essential technique in the study of patients with anemia. Its application in bone marrow aspirate smears permits the analysis of macrophage iron storage and the assessment of the number and characteristics of sideroblasts. This stain reveals ferritin granules within the erythroblasts and hemosiderin in bone marrow macrophages [7]. A proportion of normal erythroblasts exhibit few (1–5) iron-containing granules randomly distributed around the cytoplasm. Such erythroblasts are designated as sideroblasts and stand for 20–50% of the erythroblasts in a normal bone marrow [8]. Sideroblasts are visible in bone marrow aspirate smears but not in bone marrow biopsy sections since erythroblastic iron is lost in bone marrow biopsy processing [7]. Ring sideroblasts are aberrant sideroblasts where iron-loaded mitochondria are visualized by Prussian blue staining as a perinuclear ring of green-blue granules. There have been various definitions of a ring sideroblast causing confusion and controversy among clinicians [9]. The International Working Group on Morphology of Myelodysplastic Syndrome (IWGM-MDS) defined three types of sideroblasts [10]:- -
-
Type 1 sideroblasts: < 5 siderotic granules in the cytoplasm.
- -
-
Type 2 sideroblasts: ≥ 5 siderotic granules, but no perinuclear distribution.
- -
-
Type 3 or ring sideroblasts: ≥ 5 siderotic granules in a perinuclear position, covering at least one-third of the nuclear circumference.
4. Ring Sideroblast Formation
The discovery of genetic variations underlying ring sideroblast has led to a better understanding of the pathophysiology of the sideroblastic anemias. Nevertheless, ouresearchers' understanding of how ring sideroblasts arise is limited. There are many open questions in this regard: Are they detrimental to the erythroblast? Are they a cause or a consequence? Mitochondrion is the epicenter of sideroblastic anemia. Disrupted mitochondrial metabolism is present among all sideroblastic anemias for which an etiology has been defined. The mitochondrial functions affected in sideroblastic anemias are Heme biosynthesis; iron–sulfur cluster (ISC) biogenesis; and synthesis of mitochondrial proteins, general proteins, or proteins dedicated to oxidative metabolism. All of these defects lead to an aberrant accumulation of iron in the mitochondria of erythroblasts [1,12][1][12]. Figure 1 represents the main causes of acquired sideroblastic anemia.Mitochondria provide the majority of the ATP needed by eukaryotic cells through oxidative phosphorylation [13]. The adult erythrocyte is the only mammalian cell that does not have mitochondria, relying exclusively on anaerobic glycolysis for ATP production [14]. Mitochondria are semi-autonomous organelles that most likely evolved from free-floating prokaryotes that infiltrated eukaryotic cells over a billion years ago [15]. The mitochondria genome is small, around 16 kb, and replicates autonomously conserving vestiges of their prior self-sufficiency [16,17][16][17]. Mitochondrial DNA, along with several bacterial genomes, displays an intron-free circular structure [18]. Chromatin absence and a limited DNA-repair capacity enable mutations in the mitochondrial DNA to develop sideroblastic anemia [19]. Replication within mitochondria occurs independently of the nuclear genome [20]. Mitochondria are stochastically distributed to progeny after cells undergo mitosis. As a result, acquired mitochondrial abnormalities are passed on in an unequal manner to the daughter cells. This feature is important to some of the hereditary mitochondrial disorders that produce sideroblastic anemia. This characteristic also presents a conundrum regarding acquired sideroblastic anemias. Some cases of sideroblastic anemia linked with myelodysplasia include mutations that prevent some cytochromes from working properly [21,22][21][22]. It remains uncertain how mitochondria with deteriorated enzymatic performance become so prevalent in cells. Reasonably, impairment of the mitochondrion should not confer a survival advantage.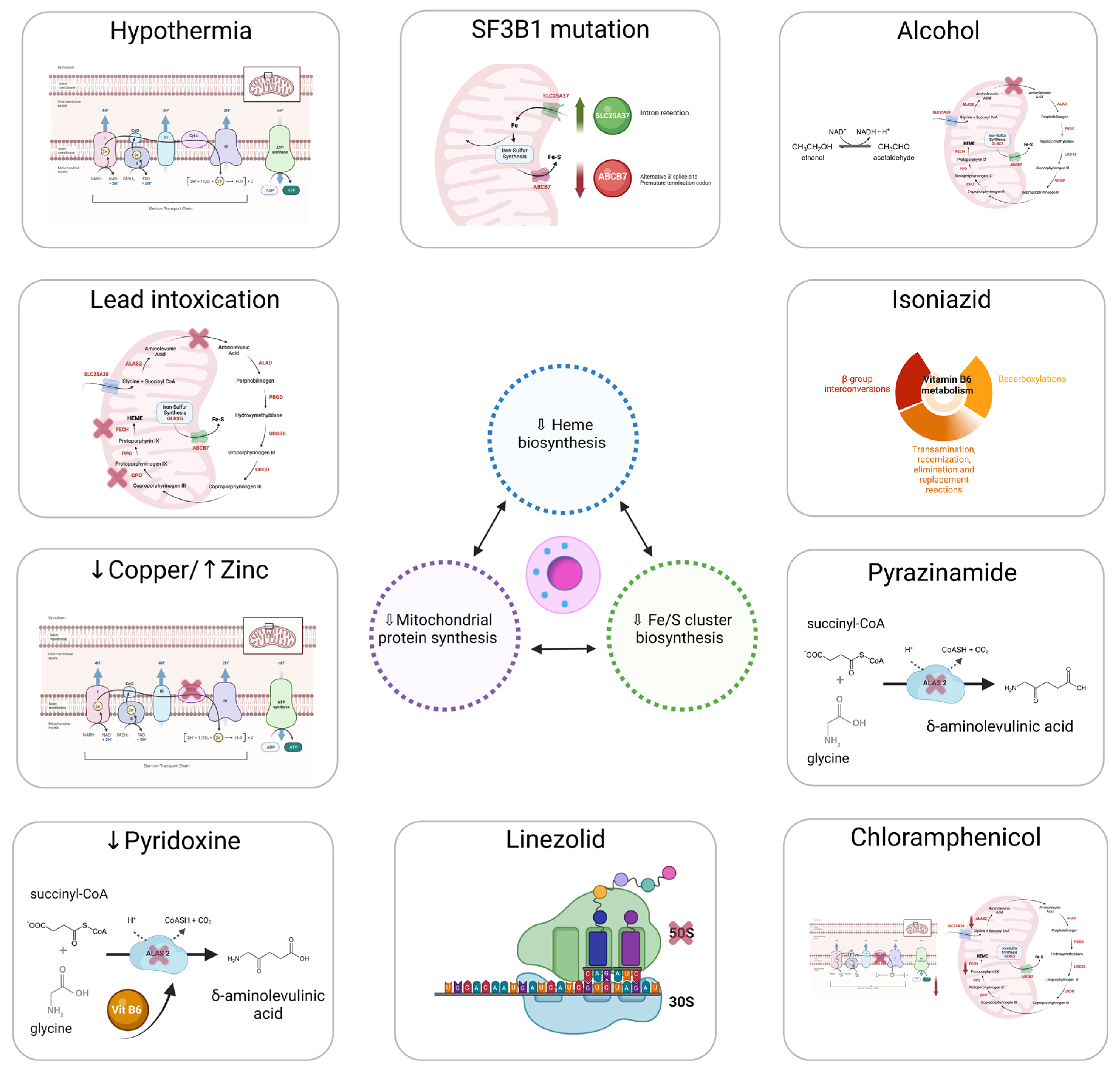 Figure 1.Main causes of acquired sideroblastic anemia.
Figure 1.Main causes of acquired sideroblastic anemia.4.1. Heme Synthesis
Most of congenital sideroblastic anemias are due to Heme deficiency. Heme is a critical component of several mitochondrial enzymes (cytochromes b, c1, c, a, and a3), as well as cytosolic enzymes such as catalase [23]. Heme plays structural and functional roles as an essential member in the hemoglobin structure. Particularly, Heme regulates the translation of globin mRNA, mediates reversible oxygen binding [24], and stabilizes the globin protein chains. Heme biosynthesis initiates with the condensation of glycine and succinyl-CoA to generate 5′-aminolevulinic acid (ALA) [25], consuming pyridoxal phosphate (active form of vitamin B6) as a cofactor in the reaction [26]. ALA then is transported to cytoplasm, where, after numerous enzymatic reactions, it is converted to coproporphyrinogen III [27]. This molecule again reaches the mitochondrion, where it undergoes further modifications and has iron inserted into the protoporphyrin IX ring by ferrochelatase (FECH), eventually generating Heme [28]. Porphyria is caused by defects in the cytoplasmic phases of Heme production. For instance, functional anomalies of the enzyme porphobilinogen deaminase produce acute intermittent porphyria [29]. Only 10 patients with erythropoietic protoporphyria (EPP) [30], a disorder characterized by pronounced deficiency of FECH, have been reported to present ring sideroblasts [31]. Aminolevulinic acid synthase (ALAS) is both the first and the rate-limiting enzyme in Heme biosynthesis [25]. Heme regulates its activity by inhibiting feedback. The two ALAS genes have been cloned and allocated to specific chromosomal regions. The ALAS-1 (also known as ALAS-n) gene has been localized to chromosome 3 (3p21) [32], being highly expressed in the liver. ALAS-1 maintains steady levels, providing basal Heme production required by all cells. ALAS-1 is a key member in the Heme biosynthetic process in mammalian cells, with the exception of erythroid cells, where erythroid-specific 5-aminolevulinate synthase (ALAS-2 or ALAS-E) governs the initial stage of Heme biosynthesis [33]. This enzyme is encoded by a gene on the X chromosome (Xp11.21), and its expression is restricted to the erythroid lineage [34]. Expression and activity of ALAS-2 is regulated by iron levels, as well as Heme-mediated feedback regulation [35]. Importantly, deficiency of ALAS-2 accounts for around 40% of all congenital sideroblastic anemia cases [36].4.2. ISC Biogenesis
Iron–sulfur clusters (ISCs) are core components of many mitochondrial and extramitochondrial proteins, showing catalytic activity [37]. ISC plays a fundamental role in cellular iron uptake regulation, iron storage, Heme synthesis, and interaction with iron regulatory protein 1 (IRP1) and FECH [38]. Congenital sideroblastic anemia, accompanied by defects in the transfer stage of ISC biogenesis, has been reported.4.3. Mitochondrial Respiratory Complex Proteins and Mitochondrial Protein Synthesis
A broad defect in mitochondrial protein synthesis has been described to lead to congenital sideroblastic anemias associated with neuromuscular disease and lactic acidosis consequent to impaired mitochondrial energy metabolism. The homeostasis of iron is vital to human health, and iron dyshomeostasis can lead to various disorders since excess iron can promote the generation of deleterious reactive oxygen species (ROS). Iron homeostasis is maintained by iron regulatory proteins (IRP1 and IRP2) and the iron-responsive element (IRE) signaling pathway [39]. Intracellular iron is used for multiple functions; if not utilized, it is stored in ferritin or exported by ferroportin in order to maintain the labile iron pool within narrow limits to avoid toxicity. Erythroblasts are cells specialized in iron uptake, and more than 80% of this iron is directed to mitochondria [40,41][40][41]. While normal erythroblasts store their iron in cytosolic ferritin, which is encoded by the FTH1 and FTL genes, ring sideroblasts store their iron in mitochondrial ferritin (FtMt), which is encoded by the FTMT gene, an intronless gene located on chromosome 5q23.1 [42]. FtMt contains ferroxidase activity; thus, it is likely to sequester potentially damaging free iron [43]. Ultimately, overexpression of FtMt results in mitochondrial iron loading and cytosolic iron deficiency [44]. The nature of this iron and the fact that these cells survive this massive overload has long been a conundrum. Bessis and Breton-Gorius found by electron microscopy that this electron dense iron gave images similar to those of the iron cores of ferritin and proposed that it was ferritin [45]. At that time, the structural complexity of ferritin was not known, and there was then no molecular basis for mitochondrial targeting. Different studies showed that there is little, if any, FtMt in normal erythroblasts but very high levels in the iron-loaded mitochondria in ringed sideroblasts [46,47][46][47]. Through Fenton chemistry (Equation (1)), iron catalyzes the creation of reactive oxygen species [48]. Molecules such as the hydroxyl radical (−OH) form in environments where oxidation processes take place around iron [49]. The mitochondrion’s oxidative metabolic machinery facilitates a suitable setting to produce reactive oxygen species. In sideroblastic anemia, the main damage that results in iron-laden mitochondria might trigger a feedback cycle of aggravating mitochondrial impairment [50]. For instance, (−OH) stimulates the peroxidation of lipids and proteins, as well as the formation of cross-links in DNA strands. Given the previously suggested lack of DNA-repair enzymes in mitochondria, the latter event might be extremely harmful. Equation (1) is the Fenton reaction. The Fenton reaction involves iron II (Fe2+) reacting with H2O2 to yield a hydroxy radical (OH) and a hydroxide ion (OH−):Fe2++H2O2→Fe3++OH.+OH−(1)65. Diagnosis of Sideroblastic Anemias
Sideroblastic anemia is primarily a laboratory diagnosis, based on the identification of ring sideroblasts in the bone marrow aspirate smear stained with the Perls’ reaction.The distinction between the different forms of sideroblastic anemia is based on the study of the characteristics of the anemia, age of diagnosis, clinical symptoms (search for symptoms suggestive of syndromic disease), and the performance of laboratory analysis, which, in many cases, involves genetic testing. Congenital sideroblastic anemias’ forms are usually diagnosed in childhood or youth and acquired forms in the elderly. However, some of the congenital sideroblastic anemias show variable expression and may be diagnosed in adulthood [51].A complete blood count (CBC), peripheral blood smear, comprehensive iron profile (e.g., ferritin, transferrin, and total iron-binding capacity (TIBC), and bonemarrow bone marrow aspiration are some of the indispensable tests that should be performed along evaluation. On the CBC, white blood cell and platelet counts are usually normal; low levels could indicate the presence of splenomegaly/hypersplenism or possible underlying causes, such as myelodysplastic syndrome (MDS). The platelet count is elevated in myeloproliferative/myelodysplastic neoplasms with ring sideroblasts and thrombocytosis (NMP/MDS-RS-T). The hemoglobin level varies between the different types of sideroblastic anemias: in inherited forms, hemoglobin tends to remain stable for long periods of time, and in MDS-RS anemia, it can be slowly progressive [51]. The mean corpuscular volume (MCV) can be a useful tool in distinguishing between the different sideroblastic anemias; most congenital sideroblastic anemias are microcytic, unlike myelodysplastic syndromes with ring sideroblasts, which usually present with macrocytic anemia [52]. The reticulocyte count is usually normal or low, which translates into ineffective erythropoiesis present in most cases.Sideroblastic anemias are characterized by a variable degree of systemic iron overload more prominently in congenital sideroblastic anemias, carrying significant morbidity and mortality. Mild-to-moderate hepatosplenomegaly is frequently seen, usually with preserved liver function. This iron overload is due to ineffective erythropoiesis, similar to what occurs in congenital dyserythropoietic anemias, thalassemia, and anemias with decreased hepcidin and increased intestinal iron absorption [51,53][51][53] In most patients with congenital sideroblastic anemias and with MDS-RS, the study of iron parameters reveals an increase in serum iron, ferritin, and transferrin saturation at diagnosis, even before the patient has required transfusion support [54].Cytological evaluation of panoptic stain shows red cells with marked anisocytosis and poikilocytosis [7]. Siderocytes and red blood cells (RBCs) in which an anomalous distribution of hemoglobin and basophilic stippling coexist are usually observed [55]. Hemosiderin particles are sometimes large and may be visible with panoptic staining (Pappenheimer bodies) [56].In the morphological study of the bone marrow aspirate, panoptic staining shows an increase in the erythroid series consisting of erythroblasts with poorly hemoglobinized cytoplasm and basophilic stippling [57]. Signs of dyserythropoiesis, such as megaloblastic changes and multinuclearity, are also seen in MDS-RS [10]. Cytoplasmic vacuolization of myeloid precursors and immature erythroid forms is common in Pearson’s syndrome [58], MLASA, and copper deficiency [8,9,10][8][9][10].To reveal ring sideroblasts, the performance of Prussian blue staining, a technique described by Max Perls in 1867, is needed. This method does not use a dye or colorant but uses hydrochloric acid to release the iron bound to proteins, and this later reacts with potassium ferrocyanide to form ferric ferrocyanide, an insoluble complex of iron with a characteristic blue-green color (Prussian blue). In sideroblastic anemias, iron retention in the macrophages is observed (in part due to intramedullary hemolysis) and the presence of ring sideroblasts (≥5 or hemosiderotic granules in a perinuclear position, covering at least one-third of the nuclear circumference) [10]. In congenital sideroblastic anemias, it is more common for ring sideroblasts to occur in late-stage erythroblasts, while in myelodysplastic syndromes, they are evident in all stages of erythroid maturation [51]. Figure 2 shows smears from a patient with MDS with ring sideroblasts.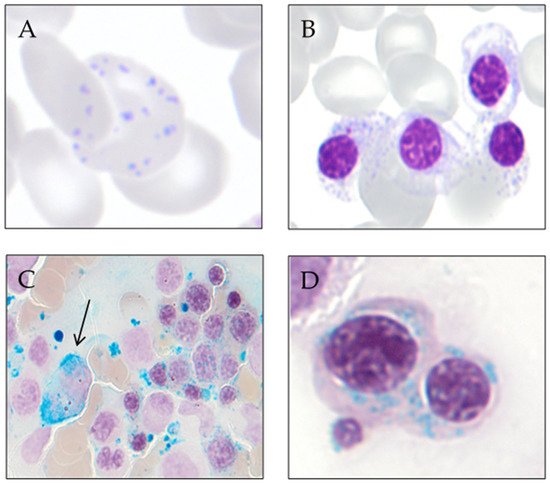 Figure 2. Smears from a patient with MDS with ring sideroblasts. (A) Peripheral blood red cell with coexistence of anomalous distribution of hemoglobin and basophilic stippling (May-Grünwald Giemsa). (B) Bone marrow erythroblast with poorly hemoglobinized cytoplasm and basophilic stippling (May-Grünwald Giemsa). (C) Bone marrow smear at low magnification showing an iron-laden macrophage (arrow) and numerous ring sideroblasts (Perls’ reaction). (D) Bone marrow ring sideroblasts (Perls’ reaction).
Figure 2. Smears from a patient with MDS with ring sideroblasts. (A) Peripheral blood red cell with coexistence of anomalous distribution of hemoglobin and basophilic stippling (May-Grünwald Giemsa). (B) Bone marrow erythroblast with poorly hemoglobinized cytoplasm and basophilic stippling (May-Grünwald Giemsa). (C) Bone marrow smear at low magnification showing an iron-laden macrophage (arrow) and numerous ring sideroblasts (Perls’ reaction). (D) Bone marrow ring sideroblasts (Perls’ reaction).
References
- Ducamp, S.; Fleming, M.D. The molecular genetics of sideroblastic anemia. Blood 2019, 133, 59–69.
- Grüneberg, H. Siderocytes: A New Kind of Erythrocytes. Nature 1941, 148, 114–115.
- Cooley, T. A severe type of hereditary anemia with elliptocytosis. Interesting sequences of splenectomy. Am. J. Med. Sci. 1945, 209, 561–568.
- Cotter, P.D.; Rucknagel, D.L.; Bishop, D.F. X-linked sideroblastic anemia: Identification of the mutation in the erythroid-specific delta-aminolevulinate synthase gene (ALAS2) in the original family described by Cooley. Blood 1994, 84, 3915–3924.
- Bjorkman, S.E. Chronic refractory anemia with sideroblastic bone marrow; a study of four cases. Blood 1956, 11, 250–259.
- Mollin, D.L. A symposium on sideroblastic anaemia held at the Postgraduate Medical School of London on Friday, March 20th, 1964, during the Annual Meeting of the British Society for Haematology. Introduction: Sideroblasts and Sideroblastic Anaemia. Br. J. Haematol. 1965, 11, 41–48.
- Swerdlow, S.H.; World Health Organization. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2017.
- Dharwadkar, A.; Vimal, S.; Panicker, N.K.; Chandanwale, S.S.; Viswanathan, V.; Kumar, H. Study of sideroblasts and iron stores in bone marrow aspirates using Perls′ stain. Med. J. Dr. DY Patil Univ. 2016, 9, 181.
- Juneja, S.K.; Imbert, M.; Jouault, H.; Scoazec, J.Y.; Sigaux, F.; Sultan, C. Haematological features of primary myelodysplastic syndromes (PMDS) at initial presentation: A study of 118 cases. J. Clin. Pathol. 1983, 36, 1129–1135.
- Mufti, G.J.; Bennett, J.M.; Goasguen, J.; Bain, B.J.; Baumann, I.; Brunning, R.; Cazzola, M.; Fenaux, P.; Germing, U.; Hellström-Lindberg, E.; et al. Diagnosis and classification of myelodysplastic syndrome: International Working Group on Morphology of myelodysplastic syndrome (IWGM-MDS) consensus proposals for the definition and enumeration of myeloblasts and ring sideroblasts. Haematologica 2008, 93, 1712–1717.
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2008; Volume 2.
- Abu-Zeinah, G.; DeSancho, M.T. Understanding Sideroblastic Anemia: An Overview of Genetics, Epidemiology, Pathophysiology and Current Therapeutic Options. J. Blood Med. 2020, 11, 305–318.
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell; Garland Science: New York, NY, USA, 2002.
- Minakami, S.; Yoshikawa, H. Studies on erythrocyte glycolysis. II. Free energy changes and rate limitings steps in erythrocyte glycolysis. J. Biochem. 1966, 59, 139–144.
- Jansen, R.P. Origin and persistence of the mitochondrial genome. Hum. Reprod. 2000, 15, 1–10.
- Zardoya, R. Recent advances in understanding mitochondrial genome diversity. F1000Research 2020, 9, 270.
- McCormick, E.M.; Muraresku, C.C.; Falk, M.J. Mitochondrial Genomics: A Complex Field Now Coming of Age. Curr. Genet. Med. Rep. 2018, 6, 52–61.
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780.
- Sulaiman, S.A.; Rani, Z.M.; Radin, F.Z.M.; Murad, N.A.A. Advancement in the diagnosis of mitochondrial diseases. J. Transl. Genet. Genom. 2020, 4, 159–187.
- Kuroiwa, T. The discovery of the division apparatus of plastids and mitochondria. QJM Int. J. Med. 2000, 49, 123–134.
- Broker, S.; Meunier, B.; Rich, P.; Gattermann, N.; Hofhaus, G. MtDNA mutations associated with sideroblastic anaemia cause a defect of mitochondrial cytochrome c oxidase. JBIC J. Biol. Inorg. Chem. 1998, 258, 132–138.
- Gattermann, N.; Retzlaff, S.; Wang, Y.L.; Hofhaus, G.; Heinisch, J.; Aul, C.; Schneider, W. Heteroplasmic point mutations of mitochondrial DNA affecting subunit I of cytochrome c oxidase in two patients with acquired idiopathic sideroblastic anemia. Blood 1997, 90, 4961–4972.
- Rascati, R.; Parsons, P. Purification and characterization of cytochrome c oxidase from rat liver mitochondria. J. Biol. Chem. 1979, 254, 1586–1593.
- Chen, J.-J. Regulation of protein synthesis by the heme-regulated eIF2α kinase: Relevance to anemias. Blood 2006, 109, 2693–2699.
- Bottomley, S.S.; Muller-Eberhard, U. Pathophysiology of heme synthesis. Semin. Hematol. 1988, 25, 282–302.
- Fujiwara, T.; Harigae, H. Biology of Heme in Mammalian Erythroid Cells and Related Disorders. BioMed Res. Int. 2015, 2015, 278536.
- Rudd, D.M. Elsevier’s Integrated Review Biochemistry, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2012.
- Ponka, P. Tissue-Specific Regulation of Iron Metabolism and Heme Synthesis: Distinct Control Mechanisms in Erythroid. Cells Blood 1997, 89, 1–25.
- Mustajoki, S.; Laine, M.; Lahtela, M.; Mustajoki, P.; Peltonen, L.; Kauppinen, R. Acute Intermittent Porphyria: Expression of Mutant and Wild-Type Porphobilinogen Deaminase in COS-1 Cells. Mol. Med. 2000, 6, 670–679.
- Rademakers, L.H.P.M.; Koningsberger, J.C.; Sorber, C.W.J.; DE LA Faille, H.B.; VAN Hattum, J.; Marx, J.J.M. Accumulation of iron in erythroblasts of patients with erythropoietic protoporphyria. Eur. J. Clin. Investig. 1993, 23, 130–138.
- Lecha, M.; Puy, H.; Deybach, J.C. Erythropoietic protoporphyria. Orphanet. J. Rare Dis. 2009, 4, 19.
- Bishop, D.F.; Henderson, A.S.; Astrin, K.H. Human delta-aminolevulinate synthase: Assignment of the housekeeping gene to 3p21 and the erythroid-specific gene to the X chromosome. Genomics 1990, 7, 207–214.
- Riddle, R.D.; Yamamoto, M.; Engel, J.D. Expression of delta-aminolevulinate synthase in avian cells: Separate genes encode erythroid-specific and nonspecific isozymes. Proc. Natl. Acad. Sci. USA 1989, 86, 792–796.
- Cotter, P.D.; Willard, H.F.; Gorski, J.L.; Bishop, D.F. Assignment of human erythroid delta-aminolevulinate synthase (ALAS2) to a distal subregion of band Xp11.21 by PCR analysis of somatic cell hybrids containing X; autosome translocations. Genomics 1992, 13, 211–212.
- Cox, T.C.; Bawden, M.J.; Martin, A.; May, B.K. Human erythroid 5-aminolevulinate synthase: Promoter analysis and identification of an iron-responsive element in the mRNA. EMBO J. 1991, 10, 1891–1902.
- Bergmann, A.K.; Bs, D.R.C.; BS, E.M.M.; Agarwal, S.; Fleming, M.D.; Bottomley, S.S.; Neufeld, E.J. Systematic molecular genetic analysis of congenital sideroblastic anemia: Evidence for genetic heterogeneity and identification of novel mutations. Pediatr. Blood Cancer 2009, 54, 273–278.
- Stehling, O.; Lill, R. The Role of Mitochondria in Cellular Iron-Sulfur Protein Biogenesis: Mechanisms, Connected Processes, and Diseases. Cold Spring Harb. Perspect. Biol. 2013, 5, a011312.
- Bottomley, S.S.; Fleming, M.D. Sideroblastic anemias: Molecular basis, pathophysiology, and clinical aspects. In Handbook of Porphyrin Science with Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine; Porphyrias and Sideroblastic Anemias, World Scientific: Singapore, 2014; Volume 29, pp. 43–87.
- Zhang, D.-L.; Ghosh, M.C.; Rouault, T.A. The physiological functions of iron regulatory proteins in iron homeostasis—An update. Front. Pharmacol. 2014, 5, 124.
- Hamdi, A.; Roshan, T.M.; Kahawita, T.M.; Mason, A.B.; Sheftel, A.D.; Ponka, P. Erythroid cell mitochondria receive endosomal iron by a “kiss-and-run” mechanism. Biochim. Biophys. Acta 2016, 1863, 2859–2867.
- Camaschella, C.; Nai, A.; Silvestri, L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 2020, 105, 260–272.
- Drysdale, J.; Arosio, P.; Invernizzi, R.; Cazzola, M.; Volz, A.; Corsi, B.; Biasiotto, G.; Levi, S. Mitochondrial Ferritin: A New Player in Iron Metabolism. Blood Cells Mol. Dis. 2002, 29, 376–383.
- Bou-Abdallah, F.; Santambrogio, P.; Levi, S.; Arosio, P.; Chasteen, N.D. Unique Iron Binding and Oxidation Properties of Human Mitochondrial Ferritin: A Comparative Analysis with Human H-chain Ferritin. J. Mol. Biol. 2005, 347, 543–554.
- Nie, G.; Sheftel, A.D.; Kim, S.F.; Ponka, P.; Brown, P.; Levis, M.; Shurtleff, S.; Campana, D.; Downing, J.; Small, D. Overexpression of mitochondrial ferritin causes cytosolic iron depletion and changes cellular iron homeostasis. Blood 2005, 105, 2161–2167.
- Bessis, M.C.; Breton-Gorius, J. Ferritin and Ferruginous Micelles in Normal Erythroblasts and Hypochromic Hypersideremic Anemias. Blood 1959, 14, 423–432.
- Levi, S.; Corsi, B.; Bosisio, M.; Invernizzi, R.; Volz, A.; Sanford, D.; Arosio, P.; Drysdale, J. A Human Mitochondrial Ferritin Encoded by an Intronless Gene. J. Biol. Chem. 2001, 276, 24437–24440.
- Cazzola, M.; Invernizzi, R.; Bergamaschi, G.; Levi, S.; Corsi, B.; Travaglino, E.; Rolandi, V.; Biasiotto, G.; Drysdale, J.; Arosio, P. Mitochondrial ferritin expression in erythroid cells from patients with sideroblastic anemia. Blood 2003, 101, 1996–2000.
- Liochev, S.I.; Fridovich, I. The role of O2.- in the production of HO.: In vitro and in vivo. Free Radic. Biol. Med. 1994, 16, 29–33.
- Gutteridge, J.M.; Rowley, D.A.; Halliwell, B. Superoxide-dependent formation of hydroxyl radicals in the presence of iron salts. Detection of ‘free’ iron in biological systems by using bleomycin-dependent degradation of DNA. Biochem. J. 1981, 199, 263–265.
- Xu, W.; Barrientos, T.; Andrews, N.C. Iron and Copper in Mitochondrial Diseases. Cell Metab. 2013, 17, 319–328.
- Bottomley, S.S.; Fleming, M.D. Sideroblastic anemia: Diagnosis and management. Hematol. Oncol. Clin. N. Am. 2014, 28, 653–670.
- Camaschella, C. Hereditary Sideroblastic Anemias: Pathophysiology, Diagnosis, and Treatment. Semin. Hematol. 2009, 46, 371–377.
- Tanno, T.; Miller, J.L. Iron Loading and Overloading due to Ineffective Erythropoiesis. Adv. Hematol. 2010, 2010, 358283.
- Ohba, R.; Furuyama, K.; Yoshida, K.; Fujiwara, T.; Fukuhara, N.; Onishi, Y.; Manabe, A.; Ito, E.; Ozawa, K.; Kojima, S.; et al. Clinical and genetic characteristics of congenital sideroblastic anemia: Comparison with myelodysplastic syndrome with ring sideroblast (MDS-RS). Ann. Hematol. 2012, 92, 1–9.
- Zahid, M.F.; Khan, N.; Pei, J.; Testa, J.R.; Dulaimi, E. Genomic imbalances in peripheral blood confirm the diagnosis of myelodysplastic syndrome in a patient presenting with non-immune hemolytic anemia. Leuk. Res. Rep. 2016, 5, 23–26.
- Woessner, S.F.L. La Citología Óptica en el Diagnóstico Hematológico, 5th ed.; Acción Médica: Madrid, Spain, 2006.
- Acín, P.; Florensa, L.; Andreu, L.; Woessner, S. Cytoplasmic abnormalities of erythroblasts as a marker for ringed sideroblasts in myelodysplastic syndromes. Eur. J. Haematol. 2009, 54, 276–278.
- Pearson, H.A.; Lobel, J.S.; Kocoshis, S.A.; Naiman, J.L.; Windmiller, J.; Lammi, A.T.; Hoffman, R.; Marsh, J.C. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J. Pediatr. 1979, 95, 976–984.