Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Rita Balsano and Version 2 by Rita Xu.
Cachexia is a metabolic syndrome consisting of massive loss of muscle mass and function that has a severe impact on the quality of life and survival of cancer patients. Up to 20% of lung cancer patients and up to 80% of pancreatic cancer patients are diagnosed with cachexia, leading to death in 20% of them. The main drivers of cachexia are cytokines such as interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), macrophage inhibitory cytokine 1 (MIC-1/GDF15) and transforming growth factor-beta (TGF-β). Besides its double-edged role as a tumor suppressor and activator, TGF-β causes muscle loss through myostatin-based signaling, involved in the reduction in protein synthesis and enhanced protein degradation.
- cachexia
- TGF-β
- cancer-related syndrome
1. Introduction
Cachexia is a multifactorial metabolic and immune system imbalance that represents one of the most detrimental side effects of cancer and anti-tumoral treatment [1]. Cancer-associated cachexia is a paraneoplastic syndrome consisting of ongoing skeletal muscle loss (with or without fat mass loss) during cancer appearance and treatment], which cannot be fully reversed by standard or enriched nutritional support, leading to progressive functional impairment and death [2]. Body emaciation, progressive loss of deambulatory function and body weight as well as an increased sense of fatigue are some of the clinical hallmarks of cancer cachexia. Half of all cancer patients develop cachexia, and this estimate increases to 80% in hospitalized or advanced-stage patients [3].
The incidence and prevalence of cancer cachexia are not homogenous across cancer patients. They rather occur at different rates depending on the type and stage of cancer. For instance, cachexia is observed in 80% of gastric, pancreatic, and esophageal cancer patients, 70% of individuals suffering from head-and-neck tumors and 60% of the combined patients with lung, colorectal, lymphoma, and prostate cancer [4]. However, cachexia is the cause of death in at least 22% of all cancer patients [5]. In addition, it has been established that cachexia can lead to lower responsiveness to anticancer therapies, worsening the quality of life of patients, and is associated with poor prognosis in advanced cancer patients [6]. With respect to the lower responsiveness, it has been reported that treating cachectic patients with conventional chemotherapeutics further enhances muscle hyper-catabolism forcing therapy discontinuation for the undesirable toxicity and could also cause detrimental changes in fat and bone mass. This would exacerbate the pathological condition, thus requiring dosage limitation or early therapy interruption [7].
Cachexia progression is often described as ranging from pre-cachexia to cachexia, and finally to refractory cachexia, where the expected survival is less than 3 months [2]. Even though its pathologic mechanisms are complex, it is often mistakenly regarded as a homogeneous condition, with little understanding that the underlying causes can be heterogeneous. Cachexia involves the loss of skeletal muscle and adipose tissue, depending in part on the grade of systemic inflammation. This muscle loss can greatly reduce the quality of life of cancer patients. Cachectic patients exhibit several other symptoms and clinicopathological alterations, such as anorexia, fatigue, anemia, early satiety, weakness, altered blood biochemistry parameters, and increased levels of inflammatory factors in various organs and tissues. The knowledge of the inflammatory changes is of extreme importance for a better understanding of the clinical picture of this syndrome [5]. Previous research suggests that systemic inflammation has a role both in the progression of cancer and of cachexia [8].
The systemic inflammation is mediated by an imbalance between pro-inflammatory and anti-inflammatory cytokines, which are normally in equilibrium. In cancer patients, this equilibrium is disturbed, which results in a dysfunctional state of both immune stimulation and suppression [9]. Cytokines function by interacting with other body tissues as well as within the tumor micro-environment itself, to generate a systemic response [10]. Hereby, cytokines contribute to mechanisms that determine the initiation, promotion, invasion and metastasis of cancer [11]. Previous work also reported that the production rate of several cytokines is associated with the prevalence of cachexia in some types of cancer [12]. The main cytokines driving cachexia are IL-6, TNF-α, TGF-β and MIC-1/GDF15 [12] MIC-1/GDF15, a member of the TGF-β superfamily, is produced in large amounts by normal and cancer cells. It acts on the feeding centers in the hypothalamus and brainstem, thereby causing anorexia and eventually cachexia [13].
Next to its role as a tumor suppressor as well as tumor activator, TGF-β has an emerging role in metabolism regulation. Acting through the SMAD2/3 pathway, it causes muscle loss through myostatin-related signaling, which is involved in the reduction in protein synthesis and in the increase in protein degradation [14]. Myostatin or GDF8 is a well-known negative regulator of muscle mass [15]. In addition, TGF-β plays a role in the mechanisms behind weight loss, muscle atrophy and fibrosis [16]. Greco and colleagues proved that anti-TGF-β antibodies, inhibiting TGF-β-based signaling, significantly improved overall survival, weight, fat mass, lean body mass, skeletal muscle proteolysis and bone mineral density of mouse models with advanced pancreatic cancer. Overall, they showed that inhibiting TGF-β could decrease the metabolic changes associated with cancer cachexia and improve overall survival [17]. Multiple studies have shown the correlation between cytokine levels and both cancer and cachexia; however, the mechanisms by which these cytokines act on the tumor and body are not completely understood.
Notably, aside from cancer, cachexia is observed in the late stages of almost every major chronic illness (such as diabetes, cardiac failure, renal failure, and chronic obstructive pulmonary disease), which underlines the need for more insights into this syndrome [18]. Despite the prevalence and severity, cachexia remains understudied, while treatment options are limited due to therapy inadequacy and inconsistency [19]. Therefore, it is essential to investigate the molecular mediators involved in the onset of cachexia to find potential therapeutic targets.
2. TGF-β Signaling Activation
Puzzlingly, TGF-β plays a dual regulation in cancer, both as a tumor suppressor and tumor enhancer. On one side, the TGF-β pathway is involved in tumor suppression, inhibiting Natural Killer (NK)-cell activity and stimulating the production of regulatory T cells (Treg) that inactivate both cytotoxic and T helper cells [20]. On the other side, TGF-β activity paradoxically promotes tumor growth by interfering with many cancer-related processes such as cell proliferation, apoptosis and epithelial-to-mesenchymal transition (EMT) [21]. In the canonical pathway, signaling is mediated by three types of receptors: TGF-β receptor I (TFβRI, also known as activin receptor-like kinase ALK5), TGF-β receptor II (TFβRII) and TGF-β receptor III (TFβRIII) [22]. TGF-β ligands bind directly to TFβRII, which phosphorylates TFβRI, which, in turn, activates SMAD proteins (SMADs) (Figure 1). SMAD2 and SMAD3 proteins are activated by ligands of TGF-β in their C-terminal serine residues, while other ligands such as BMP activate SMAD5 and SMAD8. In this process, some auxiliary proteins intervene as regulators: for example, the receptor activator SARA stabilizes both SMAD2 and SMAD3 transcription factors.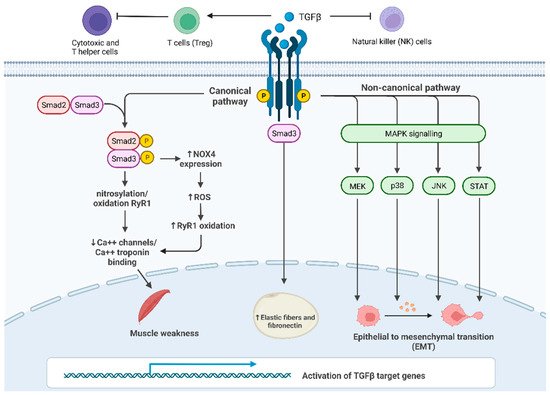
Figure 1. TGF-β signaling and its main roles in cancer progression and in cachexia: Canonical SMAD-dependent pathway: in proximal and distal skeletal muscles, SMAD3 signaling pathway results in the oxidation and nitrosylation of ryanodine receptor 1 (RyR1), which, in turn, reduces Ca2+ channels in the sarcoplasmic reticulum and causes muscle weakness; furthermore, SMAD3 induces the transcription of Nox4 gene increasing the production of ROS that oxidize RyR1. Non-canonical JNK/p38 MAPK signaling pathway affects EMT in many tissues promoting cancer growth; c. TGF-β/SMAD3 pathway leads to an increase in fibrosis in the subcutaneous adipose tissue.