1. SARS-CoV-2
Coronaviruses (CoVs) are a diverse family of enveloped positive-sense single stranded RNA viruses and consist of four genera: alpha, beta, gamma, and delta
[1][2]. Alpha and beta CoVs exclusively infect mammalian species, whereas gamma and delta CoVs are additionally capable of infecting avian species. Currently, there are seven CoV species that are known to cause human disease, referred to as human coronaviruses (HCoVs). Four of these HCoVs—HCoV-229E, HCoV-OC43, HCoV-NL63, and HCoV-HKU1—result in seasonal and mild respiratory tract infections similar to symptoms of the common cold. On the other hand, three HCoVs, i.e., Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) and Middle East Respiratory Syndrome Coronavirus (MERS-CoV), and most recently SARS-CoV-2, can result in severe, life-threatening respiratory pathologies via infection of bronchial epithelial cells, pneumocytes and upper respiratory tract cells
[2][3][1,6].
The large genome of CoVs is packed within a helical capsid formed from the nucleocapsid protein (N) and surrounded further by an envelope. The envelope is further associated with three additional structural proteins. The membrane (M) protein and the envelope (E) protein are responsible for virus assembly. The club-shaped spike (S) protein projections extend from the CoVs spherical surface and are responsible for entrance of the virus into host cells
[4][7]. The S protein consists of three segments: an ectodomain, a single-pass transmembrane anchor, and a short intracellular tail. The ectodomain further consists of a subunit responsible for receptor-binding (S1) and a subunit responsible for membrane-fusion (S2). Following attachment of the receptor binding domain (RBD) of the S1 subunit to its angiotensin-converting enzyme 2 (ACE2) receptor on the host cell surface, the S2 subunit fuses the host and viral membranes resulting in entrance of the viral genome into human cells
[1][4][2,7]. Upon entrance to the cytoplasm of the host cell, the products of replication, transcription, and translation of the viral structural proteins are assembled and released via exocytosis, allowing for the virus to continue to spread within an infected organism and to other humans via horizontal transmission
[1][2].
2. Evolving into Multiple Variants
Due to its ability to adapt to environments through mutations and recombination, and therefore altering host range and tissue tropism, SARS-CoV-2 has evolved into numerous variants referred to as variants of concern (VOCs). Evolution of CoVs is made possible due to their large genome, high mutation rate, and high recombination frequency. Mutations that result in greater fitness are selected for, resulting in VOCs. VOCs are recognized by their increased transmissibility, immuno-escape from neutralizing antibodies or T-cell immunity, and pathogenicity
[4][5][6][7,8,9]. According to the W
orld H
ealth Organization (WHO)O, to be classified as a SARS-CoV-2 variant, the virus of concern must demonstrate one or more of the following: increase in transmissibility or detrimental change in SARS-CoV-2 epidemiology, increase in virulence or change in clinical disease presentation, and/or decrease in effectiveness of public health and social measures, available diagnostics, vaccines, or therapeutics. The WHO recognizes the following five variants of SARS-CoV-2: alpha (PANGO lineage B.1.1.7), beta (PANGO lineage B1.1.351), gamma (PANGO lineage P.1), delta (PANGO lineage B.1.617.2), and most recently omicron (PANGO lineage B.1.1.529)
[7][10].
The alpha variant, which emerged out of the United Kingdom in September 2020, is understood to have functional mutations to the S protein resulting in increased transmission and increased resistance to antibody-mediated neutralization
[6][8][9,11]. First appearing in South Africa in October of 2020 is the beta variant, which has functional mutations to the S protein, similarly resulting in increased transmission
[6][9][9,12]. The gamma variant emerged out of Brazil in July of 2020 and is believed to have mutations in the RBD of the S protein resulting in increased transmission. However, less is understood about this variant
[6][10][9,13]. The delta variant, emerging out of India in October of 2020, contains mutations to the S protein resulting in increased electrostatic interactions between the RBD and ACE2 receptor (RBD-ACE2), resulting in an increase in RBD-ACE2 stability and therefore increased viral infectivity and virus replication. In addition, the delta variant is noted to have increased resistance to monoclonal antibodies, convalescent sera, and vaccinated sera, all of which have been vital treatments for CoVs infections
[6][9]. The Omicron variant, emerging out of multiple countries in November of 2021, is characterized by mutations to the S protein, similar to the Delta variant, leading to increased binding affinity between the RBD and ACE2 receptor and ultimately increased spread of the virus
[5][7][8,10]. The omicron variant has undergone further changes, leading to descendent lineages with different genetic makeup. This has led to reduction in neutralizing titers post immunization or prior infection, with currently available vaccines. However, data shows significant reduction in hospitalization rate with Omicron infection compared to the Delta variant
[7][10].
3. Increased Susceptibility to COVID-19 among Risk Groups
Although SARS-CoV-2 virus has been infectious to all age groups, regardless of their health status, age and pre-morbidities have been shown to increase the risk of adverse consequences when infected
[11][14]. Comorbidities, such as diabetes, chronic obstructive pulmonary disease (COPD), cardiovascular diseases (CVD), hypertension, malignancies, human immunodeficiency virus (HIV), and obesity, among others, have also been correlated with increased risk of severe symptom presentation when infected with SARS-CoV-2
[11][12][14,15]. Individuals with certain comorbidities highly express surface ACE2 receptors on cells
[12][15], which bind the S1 protein found on SARS-CoV-2 virus
[11][14]. Once bound, the S2 protein on SARS-CoV-2 facilitates the fusion of viral membrane with host cell membrane
[11][14]. The high release of proprotein convertase in individuals with comorbidities facilitates the endocytosis of the virus into the host cells
[12][15]. The enhanced facilitation of viral entry into host cells in individuals with comorbidities leads to significantly higher risk of morbidity and mortality
[12][15]. People suffering from diabetes mellitus (DM) and/or obesity have impaired innate and adaptive immune systems which produce more pro-inflammatory cytokines and fewer anti-inflammatory cytokines than healthy individuals
[13][16]. This suboptimal level of immunity can be further exacerbated when infected with SARS-CoV-2, leading to higher mortality and morbidity among these individuals
[13][16].
4. Pathogenesis of the Disease Induced by Cytokine Storm, Increased IL-6, Decreased Glutathione
It appears that all viruses deplete reduced glutathione (GSH), an intracellular antioxidant, as part of their strategy for replication
[14][17]. In a study designed to analyze the different pathways exploited by viruses for replication, the influenza viruses increased the production of reactive oxygen species (ROS) and decreased the production of GSH. This led to a state of oxidative stress
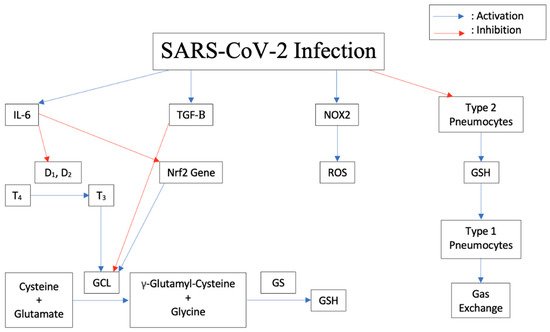
[15][16][17][18,19,20]. In response to an infection, the body releases Interleukin-6 (IL-6) and other proinflammatory cytokines. As shown in
Figure 1, IL-6 blocks the enzymes Iodothyronine deiodinases type I (D
1) and II (D
2) responsible for conversion of prohormone thyroxine T4 to active T3
[18][21]. In astrocytes, thyroid hormones upregulate glutamate cysteine ligase (GCL), which enhances GSH synthesis
[19][22]. Therefore, blocking thyroid synthesis due to a viral infection can suppress GSH synthesis and lead to increased oxidative stress in cells.
Figure 1. Downstream effects of SARS-CoV-2 Infection on cytokine release, thyroid conversion, ROS production, GSH inhibition, and pneumocyte damage in humans.
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) family consists of seven members: NOX1 to NOX5 and the two dual oxidases, Duox1 and Duox2, which are expressed in most cell types
[20][23]. The NOX family mediates ROS production in cells which plays an important role in the survival of these cells. During a viral infection, inflammatory cells release NOX2 which enhances the pathogenicity of viruses, similar to influenza A viruses
[21][24]. It has been shown that inhibition of NOX2 oxidase activity lessens influenza A virus-induced lung inflammation in mice
[22][25]. GSH acts as the main intracellular antioxidant against ROS through oxidation of the thiol group of its cysteine to a disulfide bond (GSSG), hence reducing the oxidized species and maintaining the redox state of the cell. GSSG is then converted back to GSH through the action of glutathione reductase. GSH also neutralizes the potentially harmful metals and xenobiotics in the body
[23][26]. In addition, GSH can act as a signaling molecule to the innate immune system
[23][24][25][26,27,28]. The decrease in GSH levels in infected individuals allowed viral glycoprotein haemagglutinin folding and maturation and hence viral replication
[26][29].
Elevated serum concentrations of IL-6 and TGF-β were noted in patients with severe SARS-CoV-2 symptoms compared to those with less severe symptoms
[27][30]. It has been shown that IL-6 induced a dose-dependent decrease in intracellular GSH levels in a number of human cell lines, including lung cells
[18][21]. Additionally, an animal study has shown that IL-6 can down regulate the expression of Nrf2 target genes resulting in decreased expression of the enzyme GCL, which is needed to form GSH in cells, leading to a decreased availability of GSH
[28][31]. Research showed that transforming growth factor β (TGF-β) is present in SARS-CoV-2 patients and can be classified as a strong predictor of SARS-CoV-2 disease severity
[29][32]. Notably, high levels of monocyte chemoattractant protein-1 (MCP-1) and TGF-β1 were identified in an autopsy for SARS-CoV-2 infected lung cells
[30][33]. It has been shown that TGF-β suppresses GCL gene expression and induces oxidative stress in a lung fibrosis model
[31][34]. GCL controls GSH formation intracellularly
[32][35]. The elevation of TGF-β adds additional suppression of GSH formation during SARS-CoV-2 infection. High levels of TGF-β1 may trigger fibrotic changes and may account for the typical early CT scan features of SARS-CoV-2 pneumonia such as the “ground-glass opacity” appearance
[33][34][36,37]. Fibrosis is a major complication related to SARS-CoV-2 infection
[34][35][37,38] and is related to TGF-β elevation
[31][34]. The high levels of MCP-1 explain the findings of a mononuclear inflammatory infiltrate with lymphocytes and CD68+ macrophages in the lungs of SARS-CoV-2 patients
[35][36][37][38][38,39,40,41].
Both respiratory syncytial virus (RSV) and SARS-CoV-2 infections deplete GSH and decrease the formation of the enzymes needed to form GSH in the cells they invade
[14][17], which occurs in immune cells as well as other cells such as lung Type II pneumocytes. Type II pneumocytes are responsible for the production of GSH, which is found at a very high level in the epithelial lining fluid (ELF) of the lung
[39][42]. When SARS-CoV-2 reaches the lower respiratory tract, it will bind to ACE2 on Type II pneumocytes, leading to viral infection and decreased function. When the Type II pneumocyte function is compromised by viral infection, the level of GSH in the ELF can become diminished, which is associated with compromise of lung function. This can lead to acute respiratory distress syndrome (ARDS)
[40][43], which is associated with SARS-CoV-2 infection
[41][44]. SARS-CoV-2 ARDS appears to have worse outcomes
[41][44]. Type I pneumocytes are supported by Type II pneumocytes, and damage leads to decreased function of the Type I pneumocyte pool. Damage to Type I pneumocytes leads to impaired gas exchange (1). This compromise of gas exchange with a buildup of carbon dioxide tension (Pa
CO2) blunts the brain’s response to hypoxia and leads to the presentation of individuals with “silent hypoxia.” Thus, patients with SARS-CoV-2 are described as exhibiting oxygen levels incompatible with life without dyspnea
[42][45].
5. Increased Susceptibility to TB Due to Decrease in Glutathione
Research has shown that the survival of intracellular
Mycobacterium tuberculosis (Mtb) is enhanced due to the increased uncleared free radicals and inflammatory cytokines as a result of GSH depletion
[43][46]. Therefore, immunocompromised individuals, such as those with Human immunodeficiency virus (HIV) and Type 2 Diabetes Mellitus (T2DM), are at higher risk of contracting Mtb infection
[43][46]. HIV infection is associated with an accumulation of ROS
[44][47] mediated by the envelope protein gp120
[45][48] and Tat proteins
[46][49]. Furthermore, NOX2 and NOX4-induced ROS overproduction has been reported in HIV gp120 treated astrocytes
[47][50]. An excess of ROS persists in HIV+ individuals even after successful HAART therapy and results in a depletion of GSH
[32][35]. It appears that the decrease in GSH is a result of the production of cytokines including IL-1, TNF-α and IL-17, and TGF-β
[32][35]. TGF-β interferes with the biosynthesis of GSH
[48][51]. Elevation of both IL-6 and TGF-β has also been shown to accompany the loss of GSH in HIV+ individuals and to be decreased by the administration of liposomal GSH
[49][50][52,53].
According to research, immunological abnormality plays a significant role in the increased susceptibility to Mtb infection in T2DM individuals
[51][54]. Through innate immunity, the elevated release of cytokines in diabetic individuals leads to a decrease in GSH production, and hence an increase in oxidative stress in cells
[32][35]. In addition, compared to healthy individuals, people with T2DM have lower levels of GCL, the rate-limiting enzyme in GSH synthesis
[43][46]. This leads to a significantly lower plasma GSH:GSSG ratio in diabetic patients when compared to controls
[52][55]. Consequently, ROS accumulate in cells, and have been associated with further complications of T2DM
[52][55]. Through adaptive immunity, research revealed that a decrease in Th1:Th2 cytokines can increase the susceptibility to Mtb infection in T2DM individuals
[51][54]. However, maintenance of normal GSH levels promotes Th-1 differentiation via IL-12 and IL-27 cytokines that were otherwise downregulated in immunocompromised patients
[43][46].
6. Modulation of Expression of Antioxidant Genes
Nuclear factor erythroid 2p45-related factor 2 (Nrf2) is a transcription factor, which has evolved as an oxidant-sensitive molecule that is activated and will transcriptionally stimulate a series of genes responsible for cytoprotection and detoxification
[53][56]. Nrf2 is one of the best-characterized antioxidative transcription factors with an oxidant/electrophile-sensor function
[54][57]. Under normal conditions, it forms a complex with Kelch-like ECH-associated protein 1 (KEAP1), a well-known negative regulator of Nrf2
[55][58]. Since KEAP1 serves as an adaptor protein for cullin-3-based E3 ubiquitin ligase, this dimeric Nrf2/KEAP1 complex subjects Nrf2 to constant ubiquitination and proteasomal degradation
[56][59]. In regard to their oxidant-sensing mechanisms, redox-sensitive twenty-five cysteine residues of KEAP1 were shown to play a key role in the regulation of the E3 ubiquitin ligase activity
[57][60]. Essentially, these cysteine residues are very susceptible to conjugation of a variety of ROS-inducing agents. Once conjugated, the KEAP1-mediated ubiquitination of Nrf2 is severely diminished
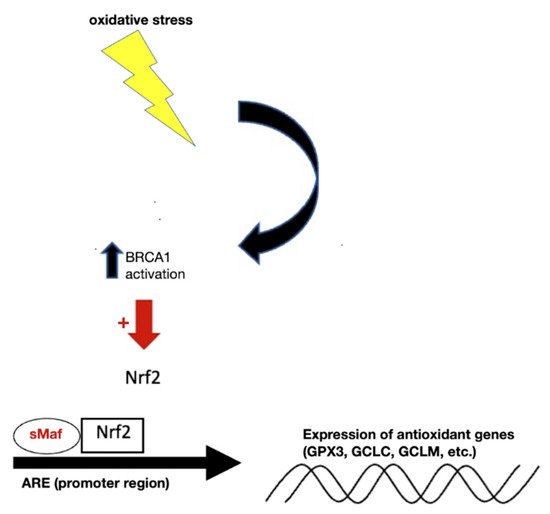
[58][61]. This leads to liberation of Nrf2 from the KEAP1-mediated restraint. Once stabilized, Nrf2 is able to get inside the nucleus and form complexes with one of small musculoaponeurotic fibrosarcoma proteins and other coactivators. As shown in
Figure 2, binding of this trimeric complex to the antioxidant response elements (AREs) in the promoter regions facilitates the transcription of a series of cytoprotective and detoxifying genes, such as heme oxygenase-1 (HO-1), NAD(P)H quinone oxidoreductase-1 (NQO-1), GCL catalytic subunit (GCLC) and GCL regulatory subunit (GCLM), glutathione S-transferase (GST), uridine diphosphate glucuronosyltransferase (UDPGT), superoxide dismutase isoform 1 (SOD1), catalase (CAT), glucose 6 phosphate dehydrogenase (G6PD), and glutathione peroxidase-1 (GPx)
[59][60][61][62][62,63,64,65]. Importantly, GCLC and GCLM are critical components of the production of GSH
[32][35].
Figure 2. Under normal conditions, BRCA1 located in the nucleus facilitates the expression of Nrf2, leading to upregulation of the ARE to produce antioxidant genes important for GSH synthesis. Abbreviations: BRCA1, breast Cancer gene 1; Nrf2, nuclear factor erythroid 2–related factor 2; ARE, antioxidant responsive element; GPX3, glutathione peroxidase 3; GCLC, glutamate-cysteine ligase catalytic subunit; GCLM, glutamate-cysteine ligase regulatory subunit.
Viruses possess a variety of adaptive mechanisms to deplete GSH in host cells. The HIV virus can decrease the expression of GCLC and GCLM in HIV+ macrophages to about half and GSH is known to be deficient in individuals with HIV
[32][63][35,66]. Respiratory Syncytial Virus (RSV) was shown to use NOX2 as an essential regulator of RSV-induced NF-κB activation. RSV virus infection led to continuous activation of NF-κB, which likely caused excessive NF-κB-mediated inflammatory gene expression
[64][67]. A later study found that RSV infection down-regulates Nrf2 expression in airway epithelial cells and that a decrease in the expression of airway antioxidant enzymes led to additional oxidative stress
[65][68]. Nrf2 mRNA levels were decreased following RSV infection and the nuclear localization of the protein was decreased in infected cells compared to uninfected ones, resulting in increased oxidation stress and a significant decrease in the GSH:GSSG ratio
[66][69].
In COVID-19 infection, depletion of GSH begins with the binding of SARS-CoV-2 S protein to ACE2, which results in inhibition and decrease of ACE2 expression in infected cells and leads to toxic overaccumulation of ANGII
[67][70]. The increased ANGII, through binding to AT1R, activates NADPH oxidases (NOX) that transfer an electron from NADPH to O
2, generating several radical species which are scavenged by GSH and deplete GSH
[67][70]. As GSH is depleted, ROS-mediated oxidation increases. Although SARS-CoV-2 proteins are synthesized in the cytosol, some viral proteins are also detectable in the nucleus, including Nsp1, Nsp5, Nsp9, Nsp13, Nsp14, and Nsp16
[68][71]. This becomes important as recent research has shown that the S protein also localizes in the nucleus and inhibits DNA damage repair by impeding key DNA repair protein BRCA1, and recruitment of 53BP1 to the damage site
[69][72].
BRCA1 is known as a tumor suppressor as mutations in this gene confer an increased risk for breast, ovarian, and prostate cancers
[70][73]. Lesser well known is BRCA1′s function in stimulating the antioxidant response element (ARE), driving transcriptional activity and its enhancement of the antioxidant response transcription factor Nrf2
[70][73]. Thus, protecting against oxidative stress is important in BRCA1′s role as a caretaker gene
[70][73]. BRCA1 increased the expression of some of these Nrf2-regulated genes (NQO1, MGST1/2, Gsta2, G6PD, and ME2), and BRCA1 also induced (and BRCA1 mutation inhibited) expression of glutathione peroxidase (GPX3)
[70][73]. In addition to endogenous reactive oxygen species, which contribute to carcinogenesis, many DNA-damaging agents and xenobiotics cause oxidative stress, resulting in DNA damage, protein oxidation, and lipid peroxidation
[71][74]. Some of these lesions are detoxified by BRCA1-regulated genes (e.g., GSTs, GPXs, oxidoreductases, and paraoxonases)
[70][73]. BRCA1 stimulates GSH production under oxidizing conditions, possibly by increasing the levels of glucose-6-phosphate dehydrogenase (G6PD). G6PD can then stimulate NADPH production, which plays a role in the regeneration of GSH
[70][73].
These findings suggest the S protein may decrease both the intracellular and intranuclear antioxidant protection of GSH. If the S protein of SARS-CoV-2 localizes to the nucleus and decreases BRCA1 recruitment and thus the benefit of Nrf2 function, evidence of oxidation of nuclear material might be evident. A decrease in available BRCA1 may contribute to the finding of increased oxidized guanine species that have been identified at higher levels in the serum of non-surviving individuals than in those surviving SARS-CoV-2
[72][75]. Additionally, it was shown that IL-6 induced a dose-dependent decrease in intracellular GSH levels in a number of human cell lines, including lung cells
[18][21]. IL-6 was shown to increase the levels of superoxide radicals, leading to the decrease of intracellular GSH
[18][21]. Increased expression of IL-6 is seen in severe SARS-CoV-2 patients, and its levels are even used to gauge the severity of the disease
[73][76]. An animal study showed that IL-6 can down regulate the expression of Nrf2 target genes resulting in decreased expression of the enzyme glutamyl cysteine ligase (GCL), leading to decreased availability of GSH
[28][31]. These findings highlight the importance of IL-6 and the S protein in maintaining a pro-inflammatory state and reducing the antioxidant protection mechanisms needed to deal with ROS.