Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Kausik Bishayee.
Alzheimer’s disease (AD) is the most common age-related dementia. The alteration in metabolic characteristics determines the prognosis. Patients at risk show reduced glucose uptake in the brain. Additionally, type 2 diabetes mellitus increases the risk of AD with increasing age. Therefore, changes in glucose uptake in the cerebral cortex may predict the histopathological diagnosis of AD.
- Alzheimer’s disease
- diabetes
- glycolysis
1. Introduction
Alzheimer’s disease (AD), the most generic form of dementia, is an irreversible, progressive brain disorder that destroys neuronal cells. AD is the fifth leading cause of death for people aged sixty-five and older [1]. Scientists do not yet fully understand the cause of this disease, which is likely to involve several factors and can affect each person differently. Health care providers often fail to diagnose AD early; thus, researchers are currently working on a diagnostic framework in which AD onset can be detected based on biological changes in the brain and body before any symptoms appear [2]. Early AD identification remains challenging as the conventional biomarkers for AD can overlap with the classical aging process. Scientists have grouped the AD biomarkers into the ATN framework, where A is amyloid, T is phosphorylated tau, and N is neurodegeneration [3]. Markers such as amyloid-β (Aβ) plaques and tau tangles are well-known indicators of AD; however, these proteins are often present at a higher physiological level in older adults [4,5,6][4][5][6].
Recent studies suggest that dysfunctional glucose metabolism is often found in AD brains. An aged-matched comparison between regular and AD brains showed reduced glucose utilization, evidenced in APP (AD model) mice [7]. Thus, glucose utilization could be an early important imaging marker for AD detection. Under normal physiological conditions, brain cells use a relatively higher percentage of glucose for their function and energy source [8]. Alterations in cerebral glucose metabolic rate and glucose consumption are reflected in the synaptic excitability and neuronal activity [9]. In the AD brain, a lesser extent of glucose utilization was detected by positron emission tomography (PET) using 18F-fluorodeoxyglucose (FDG) as a tracer [10]. Specifically, a reduction in glucose consumption at the hippocampal and posterior cingulate was observed in the early AD stages [11,12,13][11][12][13]. Later, in the advanced stages, glucose consumption reduces at the temporal-parietal cortex and the frontal and occipital cortices [12]. The decline in glucose metabolism is, moreover, correlative with synaptic density and function [14,15][14][15], suggesting that cognitive impairment has a connection to glucose consumption in the brain. The reduction in glucose consumption is directly linked with type 2 diabetes mellitus (T2DM), characterized by insulin resistance and chronic systemic inflammation. Now scientists are concentrating on the relationship between T2DM and the development of AD syndrome in the later stages of life.
2. Cerebral Glucose Metabolism
AD patients show reduced glucose metabolism in the brain. Studies have shown that improved glucose uptake and consumption have neuroprotective effects, including increased proteostasis [16]. Dysregulated glycolytic enzymes reduce glucose utilization and, alternatively, by increasing the protein glycosylation process, induce age-related neurodegeneration [17,18][17][18]. Moreover, cerebral glucose uptake in the aged brain reduces due to the reduction in the expression of the essential glucose transporters, GLUTs. For example, neuronal GLUT3 and GLUT4 expression are significantly reduced due to the lower insulin responsiveness in the T2DM brain. In addition, the glycolytic enzyme triose-phosphate isomerase (TPI) plays a crucial role in neurological dysfunction due to mutations [19]. Dysfunctional TPI is responsible for reducing the NADH content. Insufficient NADH generation reflects mitochondrial hypometabolism in TgAD mice [20]. Alternatively, inactivated TPI is also responsible for the accumulation of dihydroxyacetone phosphate (DHAP), which decomposes to methylglyoxal (MG) and is responsible for the accumulation of advanced glycation end-products (AGEs) in AD patients’ brains (Figure 1A) [21].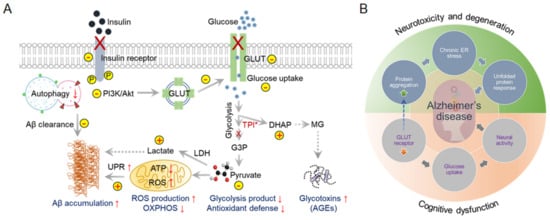
Figure 1. Dysfunctional cerebral glucose metabolism is a crucial factor in AD development. Glucose catabolism in AD is directly connected to further negative consequences in AD syndrome development. (A) Schematic representation of defective glucose metabolism in AD and various outcomes. ↑ Arrows indicate upregulation, and ↓ arrows indicate downregulation. (B) Glucose metabolism forms the bridge between neurotoxicity and cognitive dysfunction in AD. This interconnected Venn diagram describes the scenarios in which AD-related toxicity and cognitive dysfunction appear during AD onset and progression.
3. Monitoring Glucose Uptake and Metabolism in AD
The human brain uses about 20% to 25% of the total glucose utilization in the body to perform the synaptic activity. Principally, the alterations in glucose utilization practically hinder natural cellular functions, including synaptic functions in the brain. Typically, insulin receptors in brain cells control the process of glucose consumption and metabolism. Thus, the development of insulin resistance significantly raises the risk of AD [28,29][28][29]; additionally, T2DM increases the risk of AD by 50% [30,31][30][31]. In the AD brain, a lesser extent of glucose utilization was detected by positron emission tomography (PET) using 18F-fluorodeoxyglucose (FDG) as a tracer, which is a radiolabeled sugar (glucose) molecule (Figure 2). Imaging with 18F-FDG PET was used to determine the sites of abnormal glucose metabolism [32]. This detection method is now extensively used to locate tumors and sites of recurrent disease in cancer patients (ClinicalTrials.gov Identifier: NCT00207298). The use of 18F-FDG PET imaging is growing because a decrease in glucose utilization correlates with the appearance of cognitive symptoms in AD [33]. Thus, 18F-FDG PET is now considered a practical and independent biomarker for examining the metabolism changes in the pre-symptomatic phase of AD [34]. However, the use of 18F-FDG PET is still limited due to its high cost and limited availability in general clinics around the globe. Moreover, the radioactive material is unsuitable for repeated patient measurements, despite its high specificity and sensitivity.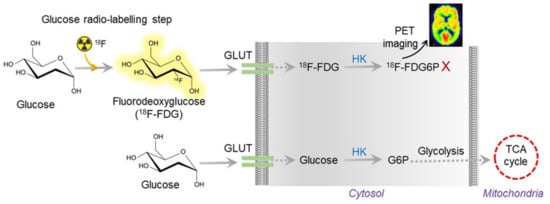
Figure 2. Overview of 18F-FDG PET working mechanism. The radioactive FDG is converted to FDG6P, which can be measured and quantified by PET imaging. In AD, glucose uptake is vastly reduced due to insulin resistance. Thus, the radioactive tracer does not show up in AD brain imaging.
References
- Alzheimer’s Association. 2022 Alzheimer’s disease facts and figures. Alzheimers Dement. 2022, 18, 700–789.
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292.
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562.
- Ebenau, J.L.; Timmers, T.; Wesselman, L.M.P.; Verberk, I.M.W.; Verfaillie, S.C.J.; Slot, R.E.R.; van Harten, A.C.; Teunissen, C.E.; Barkhof, F.; van den Bosch, K.A.; et al. ATN classification and clinical progression in subjective cognitive decline: The SCIENCe project. Neurology 2020, 95, e46–e58.
- Hojjati, S.H.; Feiz, F.; Ozoria, S.; Razlighi, Q.R.; Alzheimer’s Disease Neuroimaging, I. Topographical Overlapping of the Amyloid-beta and Tau Pathologies in the Default Mode Network Predicts Alzheimer’s Disease with Higher Specificity. J. Alzheimers Dis. 2021, 83, 407–421.
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chetelat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590.
- Huang, J.; van Zijl, P.C.M.; Han, X.; Dong, C.M.; Cheng, G.W.Y.; Tse, K.H.; Knutsson, L.; Chen, L.; Lai, J.H.C.; Wu, E.X.; et al. Altered d-glucose in brain parenchyma and cerebrospinal fluid of early Alzheimer’s disease detected by dynamic glucose-enhanced MRI. Sci. Adv. 2020, 6, eaba3884.
- Ishibashi, K.; Wagatsuma, K.; Ishiwata, K.; Ishii, K. Alteration of the regional cerebral glucose metabolism in healthy subjects by glucose loading. Hum. Brain Mapp. 2016, 37, 2823–2832.
- Mosconi, L.; Pupi, A.; De Leon, M.J. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2008, 1147, 180–195.
- Myoraku, A.; Klein, G.; Landau, S.; Tosun, D.; Alzheimer’s Disease Neuroimaging, I. Regional uptakes from early-frame amyloid PET and (18)F-FDG PET scans are comparable independent of disease state. Eur. J. Hybrid. Imaging 2022, 6, 2.
- Protas, H.D.; Chen, K.; Langbaum, J.B.; Fleisher, A.S.; Alexander, G.E.; Lee, W.; Bandy, D.; de Leon, M.J.; Mosconi, L.; Buckley, S.; et al. Posterior cingulate glucose metabolism, hippocampal glucose metabolism, and hippocampal volume in cognitively normal, late-middle-aged persons at 3 levels of genetic risk for Alzheimer disease. JAMA Neurol. 2013, 70, 320–325.
- Ferrari, B.L.; Neto, G.C.C.; Nucci, M.P.; Mamani, J.B.; Lacerda, S.S.; Felicio, A.C.; Amaro, E., Jr.; Gamarra, L.F. The accuracy of hippocampal volumetry and glucose metabolism for the diagnosis of patients with suspected Alzheimer’s disease, using automatic quantitative clinical tools. Medicine 2019, 98, e17824.
- Chen, P.; Shen, Z.; Wang, Q.; Zhang, B.; Zhuang, Z.; Lin, J.; Shen, Y.; Chen, Y.; Dai, Z.; Wu, R. Reduced Cerebral Glucose Uptake in an Alzheimer’s Rat Model With Glucose-Weighted Chemical Exchange Saturation Transfer Imaging. Front. Aging Neurosci. 2021, 13, 618690.
- Sanabria-Diaz, G.; Martinez-Montes, E.; Melie-Garcia, L.; The Alzheimer’s Disease Neuroimaging Initiative. Glucose metabolism during resting state reveals abnormal brain networks organization in the Alzheimer’s disease and mild cognitive impairment. PLoS ONE 2013, 8, e68860.
- Shivamurthy, V.K.; Tahari, A.K.; Marcus, C.; Subramaniam, R.M. Brain FDG PET and the diagnosis of dementia. Am. J. Roentgenol. 2015, 204, W76–W85.
- Duran-Aniotz, C.; Hetz, C. Glucose Metabolism: A Sweet Relief of Alzheimer’s Disease. Curr. Biol. 2016, 26, R806–R809.
- Yan, X.; Hu, Y.; Wang, B.; Wang, S.; Zhang, X. Metabolic Dysregulation Contributes to the Progression of Alzheimer’s Disease. Front. Neurosci. 2020, 14, 530219.
- Hipkiss, A.R. Aging, Alzheimer’s Disease and Dysfunctional Glycolysis; Similar Effects of Too Much and Too Little. Aging Dis. 2019, 10, 1328–1331.
- Roland, B.P.; Zeccola, A.M.; Larsen, S.B.; Amrich, C.G.; Talsma, A.D.; Stuchul, K.A.; Heroux, A.; Levitan, E.S.; VanDemark, A.P.; Palladino, M.J. Structural and Genetic Studies Demonstrate Neurologic Dysfunction in Triosephosphate Isomerase Deficiency Is Associated with Impaired Synaptic Vesicle Dynamics. PLoS Genet. 2016, 12, e1005941.
- Theurey, P.; Connolly, N.M.C.; Fortunati, I.; Basso, E.; Lauwen, S.; Ferrante, C.; Moreira Pinho, C.; Joselin, A.; Gioran, A.; Bano, D.; et al. Systems biology identifies preserved integrity but impaired metabolism of mitochondria due to a glycolytic defect in Alzheimer’s disease neurons. Aging Cell 2019, 18, e12924.
- Koike, S.; Ando, C.; Usui, Y.; Kibune, Y.; Nishimoto, S.; Suzuki, T.; Ogasawara, Y. Age-related alteration in the distribution of methylglyoxal and its metabolic enzymes in the mouse brain. Brain Res. Bull. 2019, 144, 164–170.
- Wang, Q.; Duan, L.; Li, X.; Wang, Y.; Guo, W.; Guan, F.; Ma, S. Glucose Metabolism, Neural Cell Senescence and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 4351.
- Long, D.M.; Frame, A.K.; Reardon, P.N.; Cumming, R.C.; Hendrix, D.A.; Kretzschmar, D.; Giebultowicz, J.M. Lactate dehydrogenase expression modulates longevity and neurodegeneration in Drosophila melanogaster. Aging 2020, 12, 10041–10058.
- Cai, M.; Wang, H.; Song, H.; Yang, R.; Wang, L.; Xue, X.; Sun, W.; Hu, J. Lactate Is Answerable for Brain Function and Treating Brain Diseases: Energy Substrates and Signal Molecule. Front. Nutr. 2022, 9, 800901.
- Rodic, S.; Vincent, M.D. Reactive oxygen species (ROS) are a key determinant of cancer’s metabolic phenotype. Int. J. Cancer 2018, 142, 440–448.
- Bishayee, K.; Nazim, U.M.; Kumar, V.; Kang, J.; Kim, J.; Huh, S.O.; Sadra, A. Reversing the HDAC-inhibitor mediated metabolic escape in MYCN-amplified neuroblastoma. Biomed. Pharmacother. 2022, 150, 113032.
- Sun, X.; Nie, B.; Zhao, S.; Ai, L.; Chen, Q.; Zhang, T.; Pan, T.; Wang, L.; Yin, X.; Zhang, W.; et al. Distinct relationships of amyloid-beta and tau deposition to cerebral glucose metabolic networks in Alzheimer’s disease. Neurosci. Lett. 2020, 717, 134699.
- Li, X.; Song, D.; Leng, S.X. Link between type 2 diabetes and Alzheimer’s disease: From epidemiology to mechanism and treatment. Clin. Interv. Aging 2015, 10, 549–560.
- Ferreira, L.S.S.; Fernandes, C.S.; Vieira, M.N.N.; De Felice, F.G. Insulin Resistance in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 830.
- Yu, J.H.; Han, K.; Park, S.; Cho, H.; Lee, D.Y.; Kim, J.W.; Seo, J.A.; Kim, S.G.; Baik, S.H.; Park, Y.G.; et al. Incidence and Risk Factors for Dementia in Type 2 Diabetes Mellitus: A Nationwide Population-Based Study in Korea. Diabetes Metab. J. 2020, 44, 113–124.
- Hayden, M.R. Type 2 Diabetes Mellitus Increases The Risk of Late-Onset Alzheimer’s Disease: Ultrastructural Remodeling of the Neurovascular Unit and Diabetic Gliopathy. Brain Sci. 2019, 9, 262.
- Mosconi, L.; Mistur, R.; Switalski, R.; Tsui, W.H.; Glodzik, L.; Li, Y.; Pirraglia, E.; De Santi, S.; Reisberg, B.; Wisniewski, T.; et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 811–822.
- Ou, Y.N.; Xu, W.; Li, J.Q.; Guo, Y.; Cui, M.; Chen, K.L.; Huang, Y.Y.; Dong, Q.; Tan, L.; Yu, J.T.; et al. FDG-PET as an independent biomarker for Alzheimer’s biological diagnosis: A longitudinal study. Alzheimers Res. Ther. 2019, 11, 57.
- Ricci, M.; Cimini, A.; Chiaravalloti, A.; Filippi, L.; Schillaci, O. Positron Emission Tomography (PET) and Neuroimaging in the Personalized Approach to Neurodegenerative Causes of Dementia. Int. J. Mol. Sci. 2020, 21, 7481.
- Chan, K.W.; McMahon, M.T.; Kato, Y.; Liu, G.; Bulte, J.W.; Bhujwalla, Z.M.; Artemov, D.; van Zijl, P.C. Natural D-glucose as a biodegradable MRI contrast agent for detecting cancer. Magn. Reson. Med. 2012, 68, 1764–1773.
- Sehgal, A.A.; Li, Y.; Lal, B.; Yadav, N.N.; Xu, X.; Xu, J.; Laterra, J.; van Zijl, P.C.M. CEST MRI of 3-O-methyl-D-glucose uptake and accumulation in brain tumors. Magn. Reson. Med. 2019, 81, 1993–2000.
- Kim, M.; Eleftheriou, A.; Ravotto, L.; Weber, B.; Rivlin, M.; Navon, G.; Capozza, M.; Anemone, A.; Longo, D.L.; Aime, S.; et al. What do we know about dynamic glucose-enhanced (DGE) MRI and how close is it to the clinics? Horizon 2020 GLINT consortium report. MAGMA 2022, 35, 87–104.
- Xu, X.; Yadav, N.N.; Knutsson, L.; Hua, J.; Kalyani, R.; Hall, E.; Laterra, J.; Blakeley, J.; Strowd, R.; Pomper, M.; et al. Dynamic Glucose-Enhanced (DGE) MRI: Translation to Human Scanning and First Results in Glioma Patients. Tomography 2015, 1, 105–114.
- Yadav, N.N.; Xu, J.; Bar-Shir, A.; Qin, Q.; Chan, K.W.; Grgac, K.; Li, W.; McMahon, M.T.; van Zijl, P.C. Natural D-glucose as a biodegradable MRI relaxation agent. Magn. Reson. Med. 2014, 72, 823–828.
- Rivlin, M.; Horev, J.; Tsarfaty, I.; Navon, G. Molecular imaging of tumors and metastases using chemical exchange saturation transfer (CEST) MRI. Sci. Rep. 2013, 3, 3045.
- Walker-Samuel, S.; Ramasawmy, R.; Torrealdea, F.; Rega, M.; Rajkumar, V.; Johnson, S.P.; Richardson, S.; Goncalves, M.; Parkes, H.G.; Arstad, E.; et al. In vivo imaging of glucose uptake and metabolism in tumors. Nat. Med. 2013, 19, 1067–1072.
- Tolomeo, D.; Micotti, E.; Serra, S.C.; Chappell, M.; Snellman, A.; Forloni, G. Chemical exchange saturation transfer MRI shows low cerebral 2-deoxy-D-glucose uptake in a model of Alzheimer’s Disease. Sci. Rep. 2018, 8, 9576.
More