Patients diagnosed with metastatic uveal melanoma (MUM) have a poor survival prognosis. Unfortunately for this rare disease, there is no known cure and suitable therapeutic options are limited. Histone deacetylase DAC6 inhibitors (HDAC6i) are currently in clinical trials for other cancers and show potential beneficial effects against tumor cell survival in vitro and in vivo. In MUM cells, HDAC6i show an anti-proliferative effect in vitro and in preclinical zebrafish xenograft models.
- primary and metastatic uveal melanoma
- HDAC6 inhibitors
- HDAC isozymes
- combinatorial therapies
1. Introduction
2. Selective HDAC6i as a Therapeutic Option for Uveal Melanoma
Interestingly, there are presently no registered clinical trials involving selective HDAC6i in UM/MUM, even though there are clinical trials ongoing for relapsed or refractory multiple myeloma, lymphoma, non-small cell lung cancer, metastatic breast cancer and solid tumor (Table 1) [23][29][30][31]. Likewise, to date, few studies have assessed the efficacy of selective HDAC6i in UM or MUM cells in vitro or in vivo. However, evidence in vitro and in pre-clinical UM models suggests inhibiting HDAC6 may offer therapeutic benefit. Nencetti et al. reported that novel compound VS13, with increased HDAC6 selectivity, significantly reduced 92.1 and Mel270 (primary) UM cell viability (up to a 100% reduction) in a dose-dependent manner [32]. Additionally, ACY-1215, a selective HDAC6i, induced a dose-dependent, significant reduction in UM and MUM cell survival by up to 99.99% in vitro and significantly decreased UM cell fluorescence by 65% in zebrafish xenografts with MUM (OMM2.5) cells in vivo [33].Cancer Type | Drug Combination | Clinical Trial Identifier(s) | Phase | No. of Participants | Status | Outcome | Reference (PMID) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Biliary Tract Cancer (advanced) | KA2507 | NCT04186156 EUCTR2019-001459-38-GB | Ib/II | N/A | Withdrawn | N/A | |||||||||||||||||
Relapsed/Refractory Multiple Myeloma | ACY-1215 + Bortezomib/Dexamethasone | NCT01323751 | I/II | 120 | Completed | N/A | |||||||||||||||||
Lymphoid Malignancies, Lymphoma | ACY-1215 | NCT02091063 | I/II | 24 | Completed | Under review | |||||||||||||||||
Non-Small Cell Lung Cancer | ACY-241 + Nivolumab | NCT02635061 | Ib | 17 | Active, not recruiting | Ongoing | 34552864 | ||||||||||||||||
Solid Tumor, Adult | KA2507 | NCT03008018 | I | 20 | Completed | Published | 33947698 | ||||||||||||||||
Metastatic Breast Cancer, Breast Carcinoma | ACY-1215 + Nab-paclitaxel | NCT02632071 | Ib | 17 | Completed | N/A | |||||||||||||||||
Unresectable/Metastatic Cholangiocarcinoma | ACY-1215 + Gemcitabine/Cisplatin | NCT02856568 | Ib | N/A | Withdrawn | N/A | |||||||||||||||||
Relapsed/Refractory Multiple Myeloma | ACY-1215 + Pomalidomide/Dexamethasone | EUCTR2014-002338-29-IT EUCTR2014-002338-29-GR NCT01997840 | Ib/II | 103 | Active, not recruiting | Ongoing | |||||||||||||||||
Gynecological Cancer | ACY-1215 + Paclitaxel/Bevacizumab | NCT02661815 | Ib | 6 | Terminated | Available | |||||||||||||||||
Malignant Melanoma | ACY-241 + Nivolumab + Ipilimumab | NCT02935790 | Ib | 1 | Completed | N/A | |||||||||||||||||
Solid Tumor (advanced) | JBI-802 | NCT05268666 | I/II | 126 | Recruiting | N/A |
3. Involvement of HDAC6 in Tumor Growth, Survival and Progression
Available evidence indicates HDAC6 is capable of modulating tumor growth, development, survival and progression via signaling pathways such as mitogen-activated protein kinases/extracellular-signal-regulated kinase (MAPK/ERK), phosphatidylinositol 3-kinase (PI3K/AKT) and p53 signaling cascade in cancers (Figure 1) [23][34][35][36][37][38][39]. Of these signaling pathways, MAPK/ERK and PI3K/AKT signaling, in particular, are of importance as they are implicated in UM disease pathomechanisms [40]. In the majority of UM tumors, MAPK/ERK signaling pathway is constitutively activated, which is partly attributed to mutations in genes, guanine nucleotide-binding protein G(q) subunit alpha (GNAQ) and guanine nucleotide-binding protein G(q) subunit alpha-11 (GNA11), upstream of this signaling cascade [41][42]. Uveal melanoma cells treated with a MEK and PI3K inhibitor combination inhibited cell proliferation and induced apoptosis [43]. Moreover, several other studies explored MEK and PI3K/AKT pathway inhibitors as a therapeutic option for UM; however, the therapeutic efficacy has been contentious [40][44][45][46][47].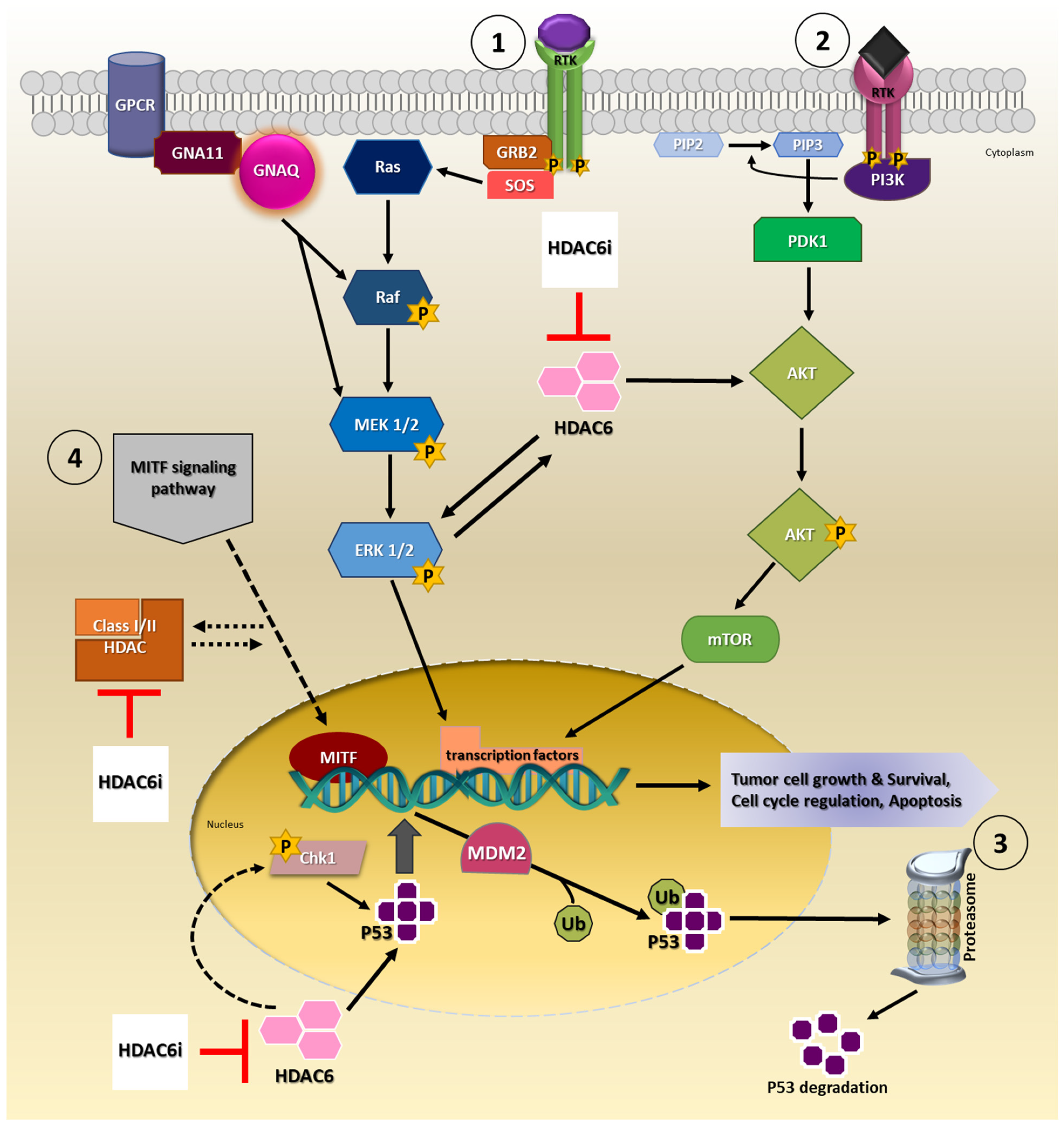
4. Immunomodulatory Effects of HDAC6 Inhibitors
The tumor microenvironment (TME) supports processes involved in tumorigenesis [75][76]. Composed of tumor cells, immune cells, stromal cells, signaling molecules, extracellular matrix and blood vessels, the microenvironment can ensure tumor cells are able to grow, survive, spread and even to gain resistance to therapy [76]. An increasing number of studies are focusing on understanding and identifying novel therapeutic targets within the tumor microenvironment for cancer treatment. In UM, tumors with chromosomal defects such as loss of one copy of chromosome 3 or gain of chromosome 8q, present with increased levels of inflammatory mediators and immune cells leading to a tumor-promoting inflammatory TME [77]. Differing from other cancers, increased amounts of tumor infiltrating lymphocytes (TILs) and tumor associated macrophages (TAMs) are correlated to poor prognosis and high metastasis risk in UM [78][79]. A delicate balance is required to ensure the prevention of UM cells from evading immune surveillance. HDACi are capable of immunomodulation in cancer [35][80][81][82][83]. Inhibition of Class I HDACs by HDACi or in combination with the immunomodulatory drug Lenalidomide, resulted in the downregulation of cellular Myc proto-oncogene protein (c-MYC) and increased cytotoxicity in multiple myeloma cells [84]. Additionally, ACY-1215 treatment, alone or combined with Lenalidomide, significantly reduced c-MYC, IKAROS family zinc finger (IKZF)1/IKZF3 and interferon regulatory factor 4 (IRF4) expression levels triggering immune system activation, which was postulated to be involved in the anti-tumor cell survival effects. The HDAC6 inhibitor A452 in combination with Lenalidomide or Pomalidomide, displayed significantly increased synergistic anti-proliferative effects which was attributed to the augmented reduction of c-MYC, IKZF1/IKZF3 and IRF4 expression in multiple myeloma cells [85]. In a preclinical mouse model of non-small cell lung cancer (NSCLC), ACY-1215 treatment increased the expression of MHC Class II molecules, CD86 and CD96 co-stimulatory molecules, suggesting that ACY-1215 may play a role in T-cell activation and antigen presentation, consequently promoting anti-tumor immunity [86]. Another study described that the HDAC6 inhibitor ACY-241 alone, and when combined with Oxaliplatin (chemotherapy drug), promoted T cell functions, thereby increasing the immunogenicity of tumor cells, in an NSCLC mouse model [87]. Inhibition of HDAC6 in melanoma cells resulted in the increased expression of MHC class I and presentation of tumor-related antigens [88]. Knox et al. demonstrated that treatment of murine melanoma model with the HDAC6i Nexturastat A, combined with the anti-PD1 antibody, increased tumor infiltration of CD8+ and natural killer cells and reduced the level of pro-tumorigenic M2 macrophages [89]. Combining HDAC6i with immunomodulatory agents can improve therapeutic efficacy [90].5. Conclusion
A combined therapeutic approach involving HDAC6i with chemotherapeutic agents, may be more beneficial and should be explored in detail as treatment options for UM/MUM. Promising results from Entinostat pembrolizumab gives us hope that it is worthwhile to pursue combinatorial treatment strategy in UM/MUM.
References
- Carvajal, R.D.; Schwartz, G.K.; Tezel, T.; Marr, B.; Francis, J.H.; Nathan, P.D. Metastatic disease from uveal melanoma: Treatment options and future prospects. Br. J. Ophthalmol. 2017, 101, 38–44.
- Branisteanu, D.C.; Bogdanici, C.M.; Branisteanu, D.E.; Maranduca, M.A.; Zemba, M.; Balta, F.; Branisteanu, C.I.; Moraru, A.D. Uveal melanoma diagnosis and current treatment options (Review). Exp. Med. 2021, 22, 1428.
- Beasley, A.B.; Chen, F.K.; Isaacs, T.W.; Gray, E.S. Future perspectives of uveal melanoma blood based biomarkers. Br. J. Cancer 2022, 126, 1511–1528.
- Kaliki, S.; Shields, C.L. Uveal melanoma: Relatively rare but deadly cancer. Eye 2017, 31, 241–257.
- Stalhammar, G.; Gill, V.T. The long-term prognosis of patients with untreated primary uveal melanoma: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2022, 172, 103652.
- Smit, K.N.; Jager, M.J.; de Klein, A.; Kili, E. Uveal melanoma: Towards a molecular understanding. Prog. Retin. Eye Res. 2020, 75, 100800.
- Rantala, E.S.; Hernberg, M.; Kivela, T.T. Overall survival after treatment for metastatic uveal melanoma: A systematic review and meta-analysis. Melanoma Res. 2019, 29, 561–568.
- Rodriguez-Vidal, C.; Fernandez-Diaz, D.; Fernandez-Marta, B.; Lago-Baameiro, N.; Pardo, M.; Silva, P.; Paniagua, L.; Blanco-Teijeiro, M.J.; Piñeiro, A.; Bande, M. Treatment of Metastatic Uveal Melanoma: Systematic Review. Cancers 2020, 12, 2557.
- Mallone, F.; Sacchetti, M.; Lambiase, A.; Moramarco, A. Molecular Insights and Emerging Strategies for Treatment of Metastatic Uveal Melanoma. Cancers 2020, 12, 2761.
- Schank, T.E.; Hassel, J.C. Immunotherapies for the Treatment of Uveal Melanoma-History and Future. Cancers 2019, 11, 1048.
- Wang, J.Z.; Lin, V.; Toumi, E.; Wang, K.; Zhu, H.; Conway, R.M.; Madigan, M.C.; Murray, M.; Cherepanoff, S.; Zhou, F.; et al. Development of new therapeutic options for the treatment of uveal melanoma. FEBS J. 2021, 288, 6226–6249.
- Schefler, A.C.; Kim, R.S. Recent advancements in the management of retinoblastoma and uveal melanoma. Fac. Rev. 2021, 10, 51.
- Nathan, P.; Hassel, J.C.; Rutkowski, P.; Baurain, J.F.; Butler, M.O.; Schlaak, M.; Sullivan, R.J.; Ochsenreither, S.; Dummer, R.; Kirkwood, J.M.; et al. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N. Engl. J. Med. 2021, 385, 1196–1206.
- Dhillon, S. Tebentafusp: First Approval. Drugs 2022, 82, 703–710.
- Agency, E.M. Kimmtrak. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/kimmtrak (accessed on 18 June 2022).
- Killock, D. Tebentafusp for uveal melanoma. Nat. Rev. Clin. Oncol. 2021, 18, 747.
- Olivier, T.; Prasad, V. Tebentafusp in first-line melanoma trials: An outperforming outlier. Transl. Oncol. 2022, 20, 101408.
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414.
- McClure, J.J.; Li, X.; Chou, C.J. Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. Adv. Cancer Res. 2018, 138, 183–211.
- Jenke, R.; Ressing, N.; Hansen, F.K.; Aigner, A.; Buch, T. Anticancer Therapy with HDAC Inhibitors: Mechanism-Based Combination Strategies and Future Perspectives. Cancers 2021, 13, 634.
- Moschos, M.M.; Dettoraki, M.; Androudi, S.; Kalogeropoulos, D.; Lavaris, A.; Garmpis, N.; Damaskos, C.; Garmpi, A.; Tsatsos, M. The Role of Histone Deacetylase Inhibitors in Uveal Melanoma: Current Evidence. Anticancer Res. 2018, 38, 3817–3824.
- Aldana-Masangkay, G.I.; Sakamoto, K.M. The role of HDAC6 in cancer. J. Biomed. Biotechnol. 2011, 2011, 875824.
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Zhang, W.; Yang, J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018, 11, 111.
- Boyault, C.; Sadoul, K.; Pabion, M.; Khochbin, S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 2007, 26, 5468–5476.
- Li, Y.; Shin, D.; Kwon, S.H. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2013, 280, 775–793.
- Batchu, S.N.; Brijmohan, A.S.; Advani, A. The therapeutic hope for HDAC6 inhibitors in malignancy and chronic disease. Clin. Sci. 2016, 130, 987–1003.
- Li, G.; Tian, Y.; Zhu, W.G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 576946.
- Hontecillas-Prieto, L.; Flores-Campos, R.; Silver, A.; de Alava, E.; Hajji, N.; Garcia-Dominguez, D.J. Synergistic Enhancement of Cancer Therapy Using HDAC Inhibitors: Opportunity for Clinical Trials. Front. Genet. 2020, 11, 578011.
- Vogl, D.T.; Raje, N.; Jagannath, S.; Richardson, P.; Hari, P.; Orlowski, R.; Supko, J.G.; Tamang, D.; Yang, M.; Jones, S.S.; et al. Ricolinostat, the First Selective Histone Deacetylase 6 Inhibitor, in Combination with Bortezomib and Dexamethasone for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2017, 23, 3307–3315.
- Amengual, J.E.; Lue, J.K.; Ma, H.; Lichtenstein, R.; Shah, B.; Cremers, S.; Jones, S.; Sawas, A. First-in-Class Selective HDAC6 Inhibitor (ACY-1215) Has a Highly Favorable Safety Profile in Patients with Relapsed and Refractory Lymphoma. Oncologist 2021, 26, 184.e366.
- Awad, M.M.; Le Bruchec, Y.; Lu, B.; Ye, J.; Miller, J.; Lizotte, P.H.; Cavanaugh, M.E.; Rode, A.J.; Dumitru, C.D.; Spira, A. Selective Histone Deacetylase Inhibitor ACY-241 (Citarinostat) Plus Nivolumab in Advanced Non-Small Cell Lung Cancer: Results from a Phase Ib Study. Front. Oncol. 2021, 11, 696512.
- Nencetti, S.; Cuffaro, D.; Nuti, E.; Ciccone, L.; Rossello, A.; Fabbi, M.; Ballante, F.; Ortore, G.; Carbotti, G.; Campelli, F.; et al. Identification of histone deacetylase inhibitors with (arylidene)aminoxy scaffold active in uveal melanoma cell lines. J. Enzym. Inhib. Med. Chem. 2021, 36, 34–47.
- Sundaramurthi, H.; Garcia-Mulero, S.; Tonelotto, V.; Slater, K.; Marcone, S.; Piulats, J.M.; Watson, R.W.; Tobin, D.J.; Jensen, L.D.; Kennedy, B.N. Uveal Melanoma Cell Line Proliferation Is Inhibited by Ricolinostat, a Histone Deacetylase Inhibitor. Cancers 2022, 14, 782.
- Zuidervaart, W.; van Nieuwpoort, F.; Stark, M.; Dijkman, R.; Packer, L.; Borgstein, A.M.; Pavey, S.; van der Velden, P.; Out, C.; Jager, M.J.; et al. Activation of the MAPK pathway is a common event in uveal melanomas although it rarely occurs through mutation of BRAF or RAS. Br. J. Cancer 2005, 92, 2032–2038.
- Cosenza, M.; Pozzi, S. The Therapeutic Strategy of HDAC6 Inhibitors in Lymphoproliferative Disease. Int. J. Mol. Sci. 2018, 19, 2337.
- Li, J.; Yu, M.; Fu, S.; Liu, D.; Tan, Y. Role of Selective Histone Deacetylase 6 Inhibitor ACY-1215 in Cancer and Other Human Diseases. Front. Pharm. 2022, 13, 907981.
- Yoo, J.; Jeon, Y.H.; Lee, D.H.; Kim, G.W.; Lee, S.W.; Kim, S.Y.; Park, J.; Kwon, S.H. HDAC6-selective inhibitors enhance anticancer effects of paclitaxel in ovarian cancer cells. Oncol. Lett. 2021, 21, 201.
- Huang, Z.; Xia, Y.; Hu, K.; Zeng, S.; Wu, L.; Liu, S.; Zhi, C.; Lai, M.; Chen, D.; Xie, L.; et al. Histone deacetylase 6 promotes growth of glioblastoma through the MKK7/JNK/c-Jun signaling pathway. J. Neurochem. 2020, 152, 221–234.
- Shoushtari, A.N.; Carvajal, R.D. GNAQ and GNA11 mutations in uveal melanoma. Melanoma Res. 2014, 24, 525–534.
- Babchia, N.; Calipel, A.; Mouriaux, F.; Faussat, A.M.; Mascarelli, F. The PI3K/Akt and mTOR/P70S6K signaling pathways in human uveal melanoma cells: Interaction with B-Raf/ERK. Investing. Ophthalmol. Vis. Sci. 2010, 51, 421–429.
- Coupland, S.E.; Lake, S.L.; Zeschnigk, M.; Damato, B.E. Molecular pathology of uveal melanoma. Eye 2013, 27, 230–242.
- Li, Y.; Shi, J.; Yang, J.; Ge, S.; Zhang, J.; Jia, R.; Fan, X. Uveal melanoma: Progress in molecular biology and therapeutics. Adv. Med. Oncol. 2020, 12, 1758835920965852.
- Khalili, J.S.; Yu, X.; Wang, J.; Hayes, B.C.; Davies, M.A.; Lizee, G.; Esmaeli, B.; Woodman, S.E. Combination small molecule MEK and PI3K inhibition enhances uveal melanoma cell death in a mutant GNAQ- and GNA11-dependent manner. Clin. Cancer Res. 2012, 18, 4345–4355.
- Faiao-Flores, F.; Emmons, M.F.; Durante, M.A.; Kinose, F.; Saha, B.; Fang, B.; Koomen, J.M.; Chellappan, S.P.; Maria-Engler, S.S.; Rix, U.; et al. HDAC Inhibition Enhances the In Vivo Efficacy of MEK Inhibitor Therapy in Uveal Melanoma. Clin. Cancer Res. 2019, 25, 5686–5701.
- Steeb, T.; Wessely, A.; Ruzicka, T.; Heppt, M.V.; Berking, C. How to MEK the best of uveal melanoma: A systematic review on the efficacy and safety of MEK inhibitors in metastatic or unresectable uveal melanoma. Eur. J. Cancer 2018, 103, 41–51.
- Farhan, M.; Silva, M.; Xingan, X.; Zhou, Z.; Zheng, W. Artemisinin Inhibits the Migration and Invasion in Uveal Melanoma via Inhibition of the PI3K/AKT/mTOR Signaling Pathway. Oxid. Med. Cell Longev. 2021, 2021, 9911537.
- Musi, E.; Ambrosini, G.; de Stanchina, E.; Schwartz, G.K. The phosphoinositide 3-kinase alpha selective inhibitor BYL719 enhances the effect of the protein kinase C inhibitor AEB071 in GNAQ/GNA11-mutant uveal melanoma cells. Mol. Cancer 2014, 13, 1044–1053.
- Chuang, M.J.; Wu, S.T.; Tang, S.H.; Lai, X.M.; Lai, H.C.; Hsu, K.H.; Sun, K.H.; Sun, G.H.; Chang, S.Y.; Yu, D.S.; et al. The HDAC inhibitor LBH589 induces ERK-dependent prometaphase arrest in prostate cancer via HDAC6 inactivation and down-regulation. PLoS ONE 2013, 8, e73401.
- Wu, J.Y.; Xiang, S.; Zhang, M.; Fang, B.; Huang, H.; Kwon, O.K.; Zhao, Y.; Yang, Z.; Bai, W.; Bepler, G.; et al. Histone deacetylase 6 (HDAC6) deacetylates extracellular signal-regulated kinase 1 (ERK1) and thereby stimulates ERK1 activity. J. Biol. Chem. 2018, 293, 1976–1993.
- Zhang, S.Z.H.; Zhou, B.; Chu, Y.; Huo, J.; Tan, Y.; Liu, D. Histone deacetylase 6 is overexpressed and promotes tumor growth of colon cancer through regulation of the MAPK/ERK signal pathway. Onco. Targets Ther. 2019, 12, 2409–2419.
- Peng, U.; Wang, Z.; Pei, S.; Ou, Y.; Hu, P.; Liu, W.; Song, J. ACY-1215 accelerates vemurafenib induced cell death of BRAF-mutant melanoma cells via induction of ER stress and inhibition of ERK activation. Oncol. Rep. 2017, 37, 1270–1276.
- Cao, J.; Lv, W.; Wang, L.; Xu, J.; Yuan, P.; Huang, S.; He, Z.; Hu, J. Ricolinostat (ACY-1215) suppresses proliferation and promotes apoptosis in esophageal squamous cell carcinoma via miR-30d/PI3K/AKT/mTOR and ERK pathways. Cell Death Dis. 2018, 9, 817.
- Katopodis, P.; Khalifa, M.S.; Anikin, V. Molecular characteristics of uveal melanoma and intraocular tumors. Oncol. Lett. 2021, 21, 9.
- Kim, C.; Lee, S.; Kim, D.; Lee, D.S.; Lee, E.; Yoo, C.; Kim, K.P. Blockade of GRP78 Translocation to the Cell Surface by HDAC6 Inhibition Suppresses Proliferation of Cholangiocarcinoma Cells. Anticancer Res. 2022, 42, 471–482.
- Kaliszczak, M.; Trousil, S.; Ali, T.; Aboagye, E.O. AKT activation controls cell survival in response to HDAC6 inhibition. Cell Death Dis. 2016, 7, e2286.
- Ellis, L.; Ku, S.Y.; Ramakrishnan, S.; Lasorsa, E.; Azabdaftari, G.; Godoy, A.; Pili, R. Combinatorial antitumor effect of HDAC and the PI3K-Akt-mTOR pathway inhibition in a Pten defecient model of prostate cancer. Oncotarget 2013, 4, 2225–2236.
- Yan, Z.; Zhang, K.; Ji, M.; Xu, H.; Chen, X. A Dual PI3K/HDAC Inhibitor Downregulates Oncogenic Pathways in Hematologic Tumors In Vitro and In Vivo. Front. Pharm. 2021, 12, 741697.
- Chilamakuri, R.; Agarwal, S. Dual Targeting of PI3K and HDAC by CUDC-907 Inhibits Pediatric Neuroblastoma Growth. Cancers 2022, 14, 1067.
- Ranganna, K.; Selvam, C.; Shivachar, A.; Yousefipour, Z. Histone Deacetylase Inhibitors as Multitarget-Directed Epi-Drugs in Blocking PI3K Oncogenic Signaling: A Polypharmacology Approach. Int. J. Mol. Sci. 2020, 21, 8198.
- Liao, W.; Yang, W.; Xu, J.; Yan, Z.; Pan, M.; Xu, X.; Zhou, S.; Zhu, Y.; Lan, J.; Zeng, M.; et al. Therapeutic Potential of CUDC-907 (Fimepinostat) for Hepatocarcinoma Treatment Revealed by Tumor Spheroids-Based Drug Screening. Front. Pharm. 2021, 12, 658197.
- Brantley, M.A., Jr.; Harbour, J.W. Deregulation of the Rb and p53 pathways in uveal melanoma. Am. J. Pathol. 2000, 157, 1795–1801.
- Helgadottir, H.; Hoiom, V. The genetics of uveal melanoma: Current insights. Appl. Clin. Genet. 2016, 9, 147–155.
- Cao, W.; Shen, R.; Richard, S.; Liu, Y.; Jalalirad, M.; Cleary, M.P.; D’Assoro, A.B.; Gradilone, S.A.; Yang, D.Q. Inhibition of triplenegative breast cancer proliferation and motility by reactivating p53 and inhibiting overactivated Akt. Oncol. Rep. 2022, 47, 1–8.
- Ryu, H.W.; Shin, D.H.; Lee, D.H.; Choi, J.; Han, G.; Lee, K.Y.; Kwon, S.H. HDAC6 deacetylates p53 at lysines 381/382 and differentially coordinates p53-induced apoptosis. Cancer Lett. 2017, 391, 162–171.
- Miyake, K.; Takano, N.; Kazama, H.; Kikuchi, H.; Hiramoto, M.; Tsukahara, K.; Miyazawa, K. Ricolinostat enhances adavosertibinduced mitotic catastrophe in TP53mutated head and neck squamous cell carcinoma cells. Int. J. Oncol. 2022, 60, 1–12.
- Dai, W.; Zhou, J.; Jin, B.; Pan, J. Class III-specific HDAC inhibitor Tenovin-6 induces apoptosis, suppresses migration and eliminates cancer stem cells in uveal melanoma. Sci. Rep. 2016, 6, 22622.
- Garraway, L.A.; Widlund, H.R.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, D.A.; Granter, S.R.; Du, J.; et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005, 436, 117–122.
- Kawakami, A.; Fisher, D.E. The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab. Investig. 2017, 97, 649–656.
- Levy, C.; Khaled, M.; Fisher, D.E. MITF: Master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006, 12, 406–414.
- Yajima, I.; Kumasaka, M.Y.; Thang, N.D.; Goto, Y.; Takeda, K.; Iida, M.; Ohgami, N.; Tamura, H.; Yamanoshita, O.; Kawamoto, Y.; et al. Molecular Network Associated with MITF in Skin Melanoma Development and Progression. J. Ski. Cancer 2011, 2011, 730170.
- Goding, C.R.; Arnheiter, H. MITF-the first 25 years. Genes Dev. 2019, 33, 983–1007.
- Gelmi, M.C.; Houtzagers, L.E.; Strub, T.; Krossa, I.; Jager, M.J. MITF in Normal Melanocytes, Cutaneous and Uveal Melanoma: A Delicate Balance. Int. J. Mol. Sci. 2022, 23, 6001.
- Phelps, G.B.; Hagen, H.R.; Amsterdam, A.; Lees, J.A. MITF deficiency accelerates GNAQ-driven uveal melanoma. Proc. Natl. Acad. Sci. USA 2022, 119, e2107006119.
- Yokoyama, S.; Feige, E.; Poling, L.L.; Levy, C.; Widlund, H.R.; Khaled, M.; Kung, A.L.; Fisher, D.E. Pharmacologic suppression of MITF expression via HDAC inhibitors in the melanocyte lineage. Pigment. Cell Melanoma Res. 2008, 21, 457–463.
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925.
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59.
- Bronkhorst, I.H.; Jager, M.J. Uveal melanoma: The inflammatory microenvironment. J. Innate Immun. 2012, 4, 454–462.
- Basile, M.S.; Mazzon, E.; Fagone, P.; Longo, A.; Russo, A.; Fallico, M.; Bonfiglio, V.; Nicoletti, F.; Avitabile, T.; Reibaldi, M. Immunobiology of Uveal Melanoma: State of the Art and Therapeutic Targets. Front. Oncol. 2019, 9, 1145.
- Tosi, A.; Cappellesso, R.; Dei Tos, A.P.; Rossi, V.; Aliberti, C.; Pigozzo, J.; Fabozzi, A.; Sbaraglia, M.; Blandamura, S.; Del Bianco, P.; et al. The immune cell landscape of metastatic uveal melanoma correlates with overall survival. J. Exp. Clin. Cancer Res. 2021, 40, 154.
- Khan, A.N.; Gregorie, C.J.; Tomasi, T.B. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol. Immunother. 2008, 57, 647–654.
- Shen, L.; Orillion, A.; Pili, R. Histone deacetylase inhibitors as immunomodulators in cancer therapeutics. Epigenomics 2016, 8, 415–428.
- Shanmugam, G.; Rakshit, S.; Sarkar, K. HDAC inhibitors: Targets for tumor therapy, immune modulation and lung diseases. Transl. Oncol. 2022, 16, 101312.
- Blaszczak, W.; Liu, G.; Zhu, H.; Barczak, W.; Shrestha, A.; Albayrak, G.; Zheng, S.; Kerr, D.; Samsonova, A.; La Thangue, N.B. Immune modulation underpins the anti-cancer activity of HDAC inhibitors. Mol. Oncol. 2021, 15, 3280–3298.
- Hideshima, T.; Cottini, F.; Ohguchi, H.; Jakubikova, J.; Gorgun, G.; Mimura, N.; Tai, Y.T.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Rational combination treatment with histone deacetylase inhibitors and immunomodulatory drugs in multiple myeloma. Blood Cancer J. 2015, 5, e312.
- Won, H.R.; Lee, D.H.; Yeon, S.K.; Ryu, H.W.; Kim, G.W.; Kwon, S.H. HDAC6selective inhibitor synergistically enhances the anticancer activity of immunomodulatory drugs in multiple myeloma. Int. J. Oncol. 2019, 55, 499–512.
- Adeegbe, D.O.; Liu, Y.; Lizotte, P.H.; Kamihara, Y.; Aref, A.R.; Almonte, C.; Dries, R.; Li, Y.; Liu, S.; Wang, X.; et al. Synergistic Immunostimulatory Effects and Therapeutic Benefit of Combined Histone Deacetylase and Bromodomain Inhibition in Non-Small Cell Lung Cancer. Cancer Discov. 2017, 7, 852–867.
- Bag, A.; Schultz, A.; Bhimani, S.; Stringfield, O.; Dominguez, W.; Mo, Q.; Cen, L.; Adeegbe, D. Coupling the immunomodulatory properties of the HDAC6 inhibitor ACY241 with Oxaliplatin promotes robust anti-tumor response in non-small cell lung cancer. Oncoimmunology 2022, 11, 2042065.
- Woan, K.V.; Lienlaf, M.; Perez-Villaroel, P.; Lee, C.; Cheng, F.; Knox, T.; Woods, D.M.; Barrios, K.; Powers, J.; Sahakian, E.; et al. Targeting histone deacetylase 6 mediates a dual anti-melanoma effect: Enhanced antitumor immunity and impaired cell proliferation. Mol. Oncol. 2015, 9, 1447–1457.
- Knox, T.; Sahakian, E.; Banik, D.; Hadley, M.; Palmer, E.; Noonepalle, S.; Kim, J.; Powers, J.; Gracia-Hernandez, M.; Oliveira, V.; et al. Selective HDAC6 inhibitors improve anti-PD-1 immune checkpoint blockade therapy by decreasing the anti-inflammatory phenotype of macrophages and down-regulation of immunosuppressive proteins in tumor cells. Sci. Rep. 2019, 9, 6136.
- Yussuf Khamis, M.; Wu, H.P.; Ma, Q.; Li, Y.H.; Ma, L.Y.; Zhang, X.H.; Liu, H.M. Overcome the tumor immunotherapy resistance by combination of the HDAC6 inhibitors with antitumor immunomodulatory agents. Bioorg. Chem. 2021, 109, 104754.