The blood-brain barrier (BBB) limits pharmacotherapy of several brain disorders. In addition to structural and metabolic characteristics of the BBB, the ATP-driven, drug efflux transporter P-glycoprotein (Pgp) is a selective gatekeeper of the BBB, and thus, a primary hindrance to drug delivery into the brain. Here we review the complex regulation of Pgp expression and functional activity at the BBB with an emphasis on recent studies from our laboratory. In addition to traditional processes such as transcriptional regulation and posttranscriptional or posttranslational modification of Pgp expression and functionality, novel mechanisms such as intra- and intercellular Pgp trafficking and intracellular Pgp-mediated lysosomal sequestration in BBB endothelial cells with subsequent disposal by blood neutrophils are discussed. These intrinsic mechanisms of active drug extrusion at the BBB are potential therapeutic targets that could be used to modulate P-glycoprotein activity in the treatment of brain diseases.
- P-glycoprotein
- ABCB1
- protein trafficking
- lysosomal sequestration
- extracellular vesicles
- neutrophils
1. Introduction
Dear author, the following contents are excerpts from your papers. They are editable.
(Due to the lack of relevant professional knowledge, our editors cannot complete a complete entry by summarizing your paper, so if you are interested in this work. you may need to write some contents by yourself. A good entry will better present your ideas, research and results to other scholars. Readers will also be able to access your paper directly through entries.)
1. Introduction
The blood-brain barrier (BBB) provides a natural defense against toxic or infective agents circulating in the blood but also restricts the brain penetration of most drugs, thus forming a bottleneck in drug development for brain diseases [1][2]. Tight junctions between brain capillary endothelial cells (BCECs) significantly reduce the permeation of small hydrophilic solutes through the intercellular cleft (paracellular pathway), thus forming a “physical barrier” [3]. The tight junctions ultimately determine the barrier properties of the BBB, but adherens junctions, which mediate the initial adhesion between endothelial cells, play a modulatory role [4]. Thus, most drugs must use predominantly transcellular pathways to reach the brain parenchyma, which is only possible in case of favorable physicochemical properties or active transport by membrane transporters of the solute carrier (SLC) family [1]. Small, lipophilic, and uncharged compounds, such as anesthetic agents, can penetrate relatively freely through the BBB by passive diffusion to reach their targets in the brain.
The blood-brain barrier (BBB) provides a natural defense against toxic or infective agents circulating in the blood but also restricts the brain penetration of most drugs, thus forming a bottleneck in drug development for brain diseases [1,2]. Tight junctions between brain capillary endothelial cells (BCECs) significantly reduce the permeation of small hydrophilic solutes through the intercellular cleft (paracellular pathway), thus forming a “physical barrier” [3]. The tight junctions ultimately determine the barrier properties of the BBB, but adherens junctions, which mediate the initial adhesion between endothelial cells, play a modulatory role [4]. Thus, most drugs must use predominantly transcellular pathways to reach the brain parenchyma, which is only possible in case of favorable physicochemical properties or active transport by membrane transporters of the solute carrier (SLC) family [1]. Small, lipophilic, and uncharged compounds, such as anesthetic agents, can penetrate relatively freely through the BBB by passive diffusion to reach their targets in the brain.
However, many of such compounds are subject to active efflux by promiscuous ATP-binding cassette (ABC) transporters such as P-glycoprotein (Pgp; MDR1; ABCB1), breast cancer resistance protein (BCRP; ABCG2) or multidrug resistance proteins (MRPs; ABCCs) that are located at the apical, blood-facing membrane of BCECs and pump drugs back into the blood before they reach the brain parenchyma [1]. As a consequence, several therapeutically important drug categories, including antiepileptic drugs (AEDs; now typically termed anti-seizure drugs [ASDs]), antidepressant drugs, anticancer drugs, or certain anti-infectious agents are restricted in their brain penetration by efflux transporters such as Pgp, which may result in the drug resistance of brain diseases [1][5][6][7]. In theory, this problem may be easily resolved by the coadministration of Pgp inhibitors. Indeed, several Pgp inhibitors or modulators have been investigated in clinical trials in patients with brain cancer or pharmacoresistant epilepsy, with limited success [2][6][8]. Several alternative strategies are actively being pursued, such as the modification of existing drugs, the development of new drugs, or the combination of novel drug delivery agents to evade Pgp-dependent efflux [9][10]. For any of these strategies, it is important to understand the transport mechanisms and regulation of ABC transporters such as Pgp at the BBB. Furthermore, the brain disease itself may alter the expression and functionality of efflux transporters at the BBB, as shown for certain types of difficult-to-treat epilepsy [2], which needs to be dealt with when developing new therapies.
However, many of such compounds are subject to active efflux by promiscuous ATP-binding cassette (ABC) transporters such as P-glycoprotein (Pgp; MDR1; ABCB1), breast cancer resistance protein (BCRP; ABCG2) or multidrug resistance proteins (MRPs; ABCCs) that are located at the apical, blood-facing membrane of BCECs and pump drugs back into the blood before they reach the brain parenchyma [1]. As a consequence, several therapeutically important drug categories, including antiepileptic drugs (AEDs; now typically termed anti-seizure drugs [ASDs]), antidepressant drugs, anticancer drugs, or certain anti-infectious agents are restricted in their brain penetration by efflux transporters such as Pgp, which may result in the drug resistance of brain diseases [1,5,6,7]. In theory, this problem may be easily resolved by the coadministration of Pgp inhibitors. Indeed, several Pgp inhibitors or modulators have been investigated in clinical trials in patients with brain cancer or pharmacoresistant epilepsy, with limited success [2,6,8]. Several alternative strategies are actively being pursued, such as the modification of existing drugs, the development of new drugs, or the combination of novel drug delivery agents to evade Pgp-dependent efflux [9,10]. For any of these strategies, it is important to understand the transport mechanisms and regulation of ABC transporters such as Pgp at the BBB. Furthermore, the brain disease itself may alter the expression and functionality of efflux transporters at the BBB, as shown for certain types of difficult-to-treat epilepsy [2], which needs to be dealt with when developing new therapies.
In order to protect the brain from intoxication, Pgp (and other efflux transporters) at the BBB should be capable of rapidly adapting their expression or activity to high levels of a potentially harmful xenobiotic in the blood (
). However, most of the adaptation processes that have been previously described are relatively slow. Thus, we hypothesized that there should be intrinsic mechanisms of active drug extrusion at the BBB that respond more rapidly to high-level drug exposure.
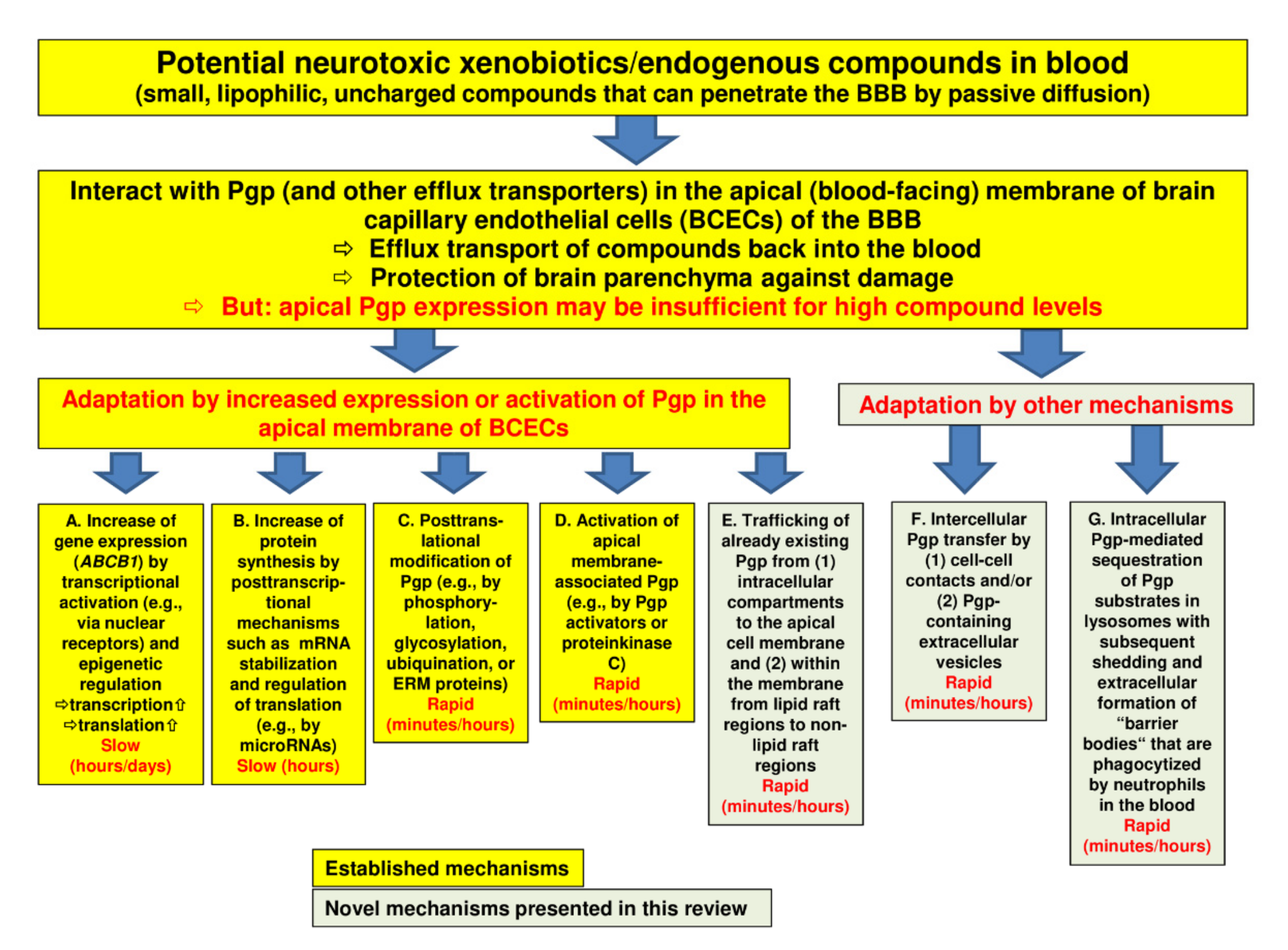
Figure 1.
Regulation of P-glycoprotein (Pgp) expression and functionality at the blood-brain barrier. The figure illustrates a number of processes by which Pgp may adapt to high blood levels of xenobiotics with the aim to protect the brain parenchyma from intoxication. The yellow boxes (designated by (
A
–
C)) show well-known mechanisms of Pgp adaptation, whereas the greenish boxes (
)) show well-known mechanisms of Pgp adaptation (see Section 2), whereas the greenish boxes (
E
–
G) illustrate the novel mechanisms that we characterized in brain capillary endothelial cells in recent years. Note that the mechanisms described in boxes (
) illustrate the novel mechanisms that we characterized in brain capillary endothelial cells in recent years (see Section 3). Note that the mechanisms described in boxes (
A
,
B
,
F
) will increase the Pgp content of the cell, whereas the mechanisms described in boxes (
C
,
D
,
E
) and (
G) will not. Although Pgp is the best characterized drug efflux transporter at the blood-brain barrier (BBB), other transporters such as breast cancer resistance protein (BCRP) and multidrug resistance proteins (MRPs) are thought to support Pgp in its role to protect the brain. Indeed, the role of these other transporters is often underestimated, particularly in older studies.
) will not. Although Pgp is the best characterized drug efflux transporter at the blood-brain barrier (BBB), other transporters such as breast cancer resistance protein (BCRP) and multidrug resistance proteins (MRPs) are thought to support Pgp in its role to protect the brain (see Section 2.6). Indeed, the role of these other transporters is often underestimated, particularly in older studies.
2. Regulation of Pgp at the BBB
The vast majority of previous studies that tried to enhance drug delivery to the brain by interfering with Pgp at the BBB used drugs that directly inhibit Pgp. In the ensuing 30 years, three distinct generations of Pgp inhibitors have been developed [6][11]. First-generation inhibitors, including verapamil, quinidine, amiodarone, and cyclosporine A, were not selective, not potent, or were toxic. The second-generation agents valspodar (PSC833) and dexverapamil were more potent but interfered with drug metabolism. The third generation of inhibitors, including dofequidar, zosuquidar, tariquidar, elacridar, and biricodar, were developed specifically as Pgp inhibitors; they were more potent and displayed fewer pharmacokinetic interactions than inhibitors of previous generations but caused toxicity in combination with chemotherapy, which was potentially due to the inhibition of Pgp expressed in normal tissue [6]. Furthermore, at higher concentrations, tariquidar and elacridar also inhibit BCRP [12]. Using PET imaging, tariquidar was shown to increase brain levels of Pgp substrates such as verapamil or d-loperamide in rodents, nonhuman primates, and humans, thus demonstrating the utility of transport inhibition at the BBB [6]. However, because of several negative clinical cancer trials with such Pgp inhibitors, interest in investigating these drugs for enhancing drug delivery to the brain has waned.
The vast majority of previous studies that tried to enhance drug delivery to the brain by interfering with Pgp at the BBB used drugs that directly inhibit Pgp. In the ensuing 30 years, three distinct generations of Pgp inhibitors have been developed [6,198]. First-generation inhibitors, including verapamil, quinidine, amiodarone, and cyclosporine A, were not selective, not potent, or were toxic. The second-generation agents valspodar (PSC833) and dexverapamil were more potent but interfered with drug metabolism. The third generation of inhibitors, including dofequidar, zosuquidar, tariquidar, elacridar, and biricodar, were developed specifically as Pgp inhibitors; they were more potent and displayed fewer pharmacokinetic interactions than inhibitors of previous generations but caused toxicity in combination with chemotherapy, which was potentially due to the inhibition of Pgp expressed in normal tissue [6]. Furthermore, at higher concentrations, tariquidar and elacridar also inhibit BCRP [183]. Using PET imaging, tariquidar was shown to increase brain levels of Pgp substrates such as verapamil or d-loperamide in rodents, nonhuman primates, and humans, thus demonstrating the utility of transport inhibition at the BBB [6]. However, because of several negative clinical cancer trials with such Pgp inhibitors, interest in investigating these drugs for enhancing drug delivery to the brain has waned.
We discuss specific strategies for interfering with Pgp activity at the BBB. The example of seizure-induced overactivity of Pgp at the BBB illustrates that interfering with the signaling pathway leading to Pgp induction is much more selective and tissue-specific than the mere direct inhibition of Pgp throughout the body. Some of the drugs involved in this pathway, e.g., COX-2 inhibitors, are clinically available and have been shown to increase the penetration of Pgp substrates into the epileptic brain. However, most of the other novel strategies discussed here are only based on in vitro findings and need to be confirmed in in vivo models for subsequent translation to the clinic. Nevertheless, continuing to uncover the regulation of Pgp at the BBB has the potential to radically change the way that we facilitate drug penetration across the BBB in the treatment of brain diseases.