Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by John Morser and Version 2 by Amina Yu.
Chemerin is the product of the RARRES2 gene which is secreted as a precursor of 143 amino acids. That precursor is inactive, but proteases from the coagulation and fibrinolytic cascades, as well as from inflammatory reactions, process the C-terminus of chemerin to first activate it and then subsequently inactivate it. Chemerin can signal via two G protein-coupled receptors, chem1 and chem2, as well as be bound to a third non-signaling receptor, CCRL2. Chemerin is produced by the liver and secreted into the circulation as a precursor, but it is also expressed in some tissues where it can be activated locally.
- chemerin
- proteases
- obesity
1. Chemerin System of Genes, Proteins, and Receptors
In a study identifying genes by subtractive hybridization in skin graft cultures that responded to treatment with a synthetic retinoid, tazarotene, a novel mRNA was identified that was named TAZAROTENE-INDUCED GENE 2 (TIG-2) [1]. HereThin, its study did not identify the role of the encoded protein, but to our knowledge this is the earliest report of the chemerin protein and gene.
An orphan receptor now known as chemerin receptor 1 (chem1, also known as chemokine-like receptor 1, CMKLR1; chemR23; DEZ) was identified as a receptor for TIG-2 protein, renamed as chemerin (also known by its gene name, retinoic acid receptor response protein 2, RARRES2) in studies in which biological fluids were evaluated for activity on cells overexpressing chem1, and those with high activity were purified through a multi-step procedure to yield a single protein. When that purified protein was analyzed by tryptic digestion, the sequence was identical to the protein encoded by the RARRES2 gene (TIG2) except for missing a few amino acids at the C-terminus [2][3][4][2,3,4].
The sequence of chemerin consists of a signal peptide allowing secretion, a core with 3 disulfide bonds, and N- and C-terminal tails (Figure 1a). No 3D crystal structure has yet been reported, although a preliminary NMR structure with most residues assigned but lacking the C-terminus is available [5]. A model is available in alphafold, predicting that there is a central domain with a relatively rigid structure containing the three disulfide bonds, while the N- and C-termini do not have tight rigid features (Figure 1b). This central domain has some structural similarities with the cathelicidin family of anti-microbial peptides as well as with the cystatin family of cysteine protease inhibitors [2][6][2,6]. It was clear from the first reports describing chemerin that the full-length protein needed to be proteolytically cleaved within the C-terminal tail to generate activity [2][4][7][2,4,7], and forms that ended at serine 157 were suggested to be the most active on the chemerin receptor, chem1. Forms that have been identified either from in vivo biopsy samples or from in vitro enzymatic studies are shown in Figure 1a.
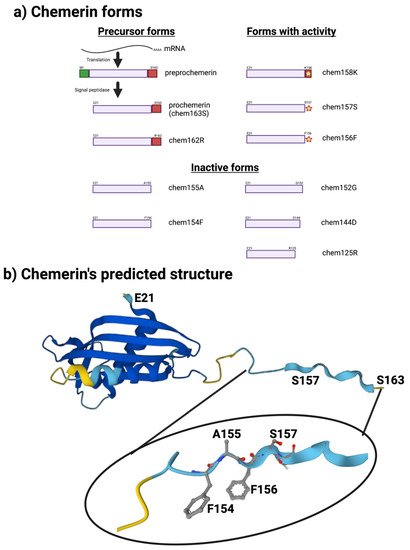
Figure 1. Chemerin forms and chemerin’s predicted structure. Created with BioRender.com. (a) Chemerin forms that have been identified in biopsy samples are grouped into those that are precursors with no intrinsic activity but can be activated by appropriate proteolysis, those that are active, and those are inactive. (b) The structure of chemerin predicted by AlphaFold [8][9][15,16] with the darker blue colors representing higher per-residue confidence in the prediction with a magnification of the C-terminal tail.
Upon binding chemerin, chem1 signals via the Gi/o family of G proteins, thereby inhibiting production of cyclic AMP while increasing production of IP3 and calcium mobilization, in addition to the activation of phospholipase C and the PI3 kinase and MAPK pathways [10][8]. Stimulation of chem1 can also activate the RhoA/ROCK pathway [11][9] and induce binding of arrestins to chem1 [12][13][10,11], which can both terminate GPCR signaling and function as a framework for assembling signaling complexes [14][12].
Chemerin has been linked to two additional receptors, chemerin receptor 2 (chem2, also known as GPR1) and chemokine receptor-like 2 (CCRL2). The nomenclature of chemerin receptors used here is that determined by the International Union of Basic and Clinical Pharmacology Committee on Receptor Nomenclature and Drug Classification based on the receptor’s cognate ligand [15][13]. Both chem1 and chem2 are members of subclass A8 belonging in the rhodopsin class of G protein-coupled receptors (GPCRs). CCRL2 does not encode a full serpentine G protein-coupled receptor GPCR and is not a signaling receptor but binds chemerin and can present it to the signaling receptors chem1 and chem2 [16][14]. The three receptors and chemerin together comprise the chemerin system.
2. Chemerin System Evolution
The chemerin gene (RARRES2) is found in all tetrapods that have been sequenced but there is no gene annotation for chemerin in fish (Figure 2). The gene consists of 6 exons of which 5 encode the protein sequence. The pattern of introns and exons as well as the encoded protein sequences have been conserved suggesting that chemerin has a significant role in tetrapod biology. In particular the C-terminal tail, responsible for binding to its receptors, chem1 and chem2, has been conserved. The gene has four exons, with the first exon encoding the signal sequence and most of the N-terminal tail, the central core being encoded by the other three exons, with the last exon encoding the final part of the central core and the C-terminal tail. In humans, the RARRES2 locus is on chromosome 7 on the antisense strand and on chromosome 6 in mice.
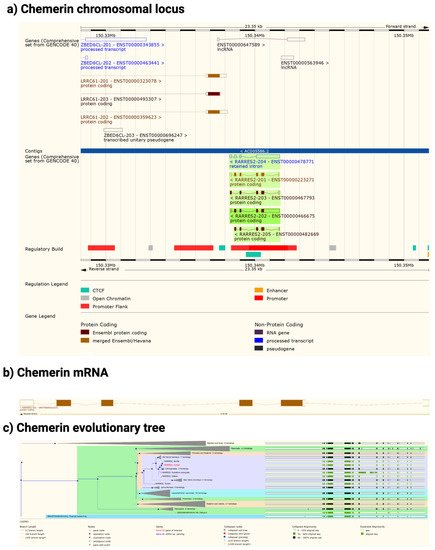
Figure 2. Chemerin gene (Rarres2), mRNA, and evolutionary tree. (a) Chromosomal neighborhood of the chemerin gene on chromosome 7. Chemerin in green on anti-sense strand. (b) Chemerin mRNA with 6 exons in boxes, filled if coding. (c) Evolutionary tree for the chemerin gene. Figures created from data in EMSEMBL human genome build GRCH38.p13 (https://ensembl.org/Homo_sapiens accessed on 11 August 2022).
In both humans and mice there is a coding gene on the sense strand, leucine rich repeat containing 61 (LRRC61) with it 3′ terminus close to the 3′ terminus of chemerin. In humans but not mice there is also an lncRNA present in the middle of the chemerin locus on the sense strand as well as a pseudogene, ZBED6CL that overlaps with the LRRC61 gene (Figure 2a). In both humans and mice five alternative spliced chemerin mRNAs have been described, but in both species there is one major coding form.
Chem1 and chem2 are similar to each other and belong to the same clade within the GPCR family, clade A8, which includes receptors for small peptides such as bradykinin and apelin. Both chem1 and chem2 genes can be identified not just in tetrapods but also in bony fish, and chem1 can be found additionally in cartilaginous fish. In addition to chemerin, chem1 and chem2 have alternative ligands for these receptors such as the resolvins E1 and E2 [17][18][19][17,18,19], and they may be a clearance receptor for amyloid beta in Alzheimer’s disease [20].
The chemerin system has been studied in model organisms such as mice and rats, as well as humans. In addition, there are data on the chemerin system in cattle, pigs, and chickens [21].
3. Chemerin mRNA Regulation
Although the chemerin gene was originally identified as being responsive to a retinoic acid receptor ligand, tazarotene [1], there have been few follow-up studies on the chemerin promoter and control of chemerin mRNA expression. The major regulatory elements controlling constitutive expression were shown to lie in the region between −252 and +258 bp around the start of mRNA transcription at +1 bp [22]. Differences in responses to IL-1b and oncostatin-M were found between hepatocytes and white adipocytes in which there was increased transcription at the chemerin promoter in adipocytes but not in hepatocytes. Those changes correlated with changes in methylation, and the list of transcription factors predicted to interact with the chemerin promoter also differed between the cell types.
4. Different Forms of the Chemerin Protein
The human chemerin cDNA encodes an open reading frame of 163 amino acids translated into preprochemerin (Figure 1 and Figure 3a), of which the first 20 comprise the signal peptide which is removed upon secretion to generate the 143 amino acid protein terminating at Ser163 [23]. When it was isolated from human ascitic fluid secondary to ovarian or liver cancer, the analysis of chemerin protein by tryptic digestion followed by mass spectrometry showed that the protein’s C-terminus was at Ser157. This was different from the C-terminus predicted from its encoding cDNA, which extends a further 6 amino acids to Ser163 [2]. Purified chemerin proteins from hemofiltrate were also found to be truncated compared to the full-length protein and were identified as terminating at Phe154 [3].
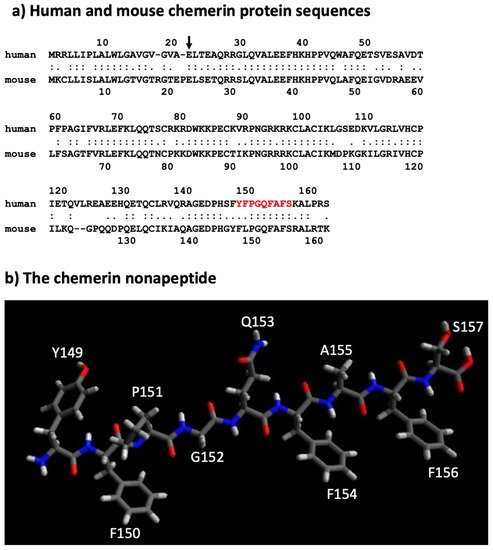
Figure 3. Sequences of mouse and human chemerin and structure of nonapeptide. (a) Mouse and human chemerin protein sequences. The arrow marks the site of cleavage by the signal peptidase, and the amino acid residues highlighted in red represent the nonapeptide. (b) chemerin nonapeptide.
In subsequent studies on serum and plasma, the major form present was found to be the precursor, chem163S. The liver produces and secretes the precursor into the circulation, shown by the treatment of rats with an anti-sense oligonucleotide that blocks chemerin synthesis specifically in the liver. The anti-sense treatment resulted in circulating levels of chem163S being substantially reduced on Western blots [24].
All forms of chemerin bind to glycosaminoglycans, such as heparin, which has been used as a one-step affinity purification to clean up samples [25]. Therefore, it would be expected that there would be a pool of chemerin bound to the glycosaminoglycans in the extracellular matrix in tissues and on blood vessels. There are no data available on this possibility nor on the pharmacokinetics of chemerin, even in reports in which mice have been treated with chemerin.
Subsequently, a variety of chemerin forms have been produced by either direct expression of them or proteolytic cleavage of a purified longer form, allowing comparative studies of the different forms. These studies have described the activity of the different forms in detail and have clarified the relative activities of the different forms on chem1 using either chemotaxis or calcium transients as the readout [26][27][26,27].
Unfortunately, similar detailed studies defining the activities of different chemerin forms are not available for chem2, although the signaling of chem1, chem2, and CCRL2 has been compared [11][12][9,10]. The results suggest that chem157S binds to chem1 with a KD of 0.88 nM and to chem2 with a KD of 0.21 nM, but signaling through chem2 may be more biased towards β-arrestin pathways than G protein pathways while the Rho/ROCK and MAPK/ERK pathways may be equivalently activated by the two receptors [28]. Although dimerization of GPCRs into homo- and hetero-dimers and maybe higher oligomers can occur, the possibility of dimerization of chem1 and chem2 into either homo- or hetero-dimers and any consequent effects on signaling have not been investigated [29][30][29,30].
A nonapeptide equivalent to amino acids 148–157 (sequence in red in Figure 3a and depicted in Figure 3b) is a very weak competitor of the binding of chem157S to chem1 or chem2 but is an effective competitor for itself. In contrast, this nonapeptide possessed good activity at chem1, almost equivalent to that of the protein with a C-terminus at amino acid Ser157 (chem157S) [26]. Carefully characterized chem157S prepared from mammalian cells gave an EC50 of low nM on chem1, with the difference in activity between the nonapeptide and chem157S being ~3-fold [26][27][26,27]. In assays in which the G protein signaling pathways were analyzed, this nonapeptide had greatly reduced activity compared to chem157S.
This data suggests that most, if not all, of the binding energy between chemerin and chem1 was located in that C-terminal tail terminating at Ser157. Further truncations of the peptides resulted in a loss of binding and activity, highlighting the importance of the C-terminal serine at position 157 [26]. Also important is Phe156, the penultimate residue in the active nonapeptide, based on the activity of an alanine substitution at that position as well as removal of the C-terminal Ser157.
Accurate determination of binding constants and activity values such as KD and EC50 for different chemerin forms or peptides requires the preparation of several independent lots of the protein or peptide, followed by an analysis of purity by methods allowing demonstration that the samples are a single molecular entity comprising a monomolecular solution of known and high purity and concentration. These independent lots can then be assayed multiple times in order to accurately estimate the KD and EC50. Most preparations of chemerin proteins have not been analyzed by methods that would allow detection of aggregates, such as non-reducing SDS-PAGE or size exclusion chromatography. These analyses are especially critical for E. coli-produced material as chemerin is commonly produced as an inclusion body requiring solubilization and refolding. Evaluation of the integrity and purity of chemerin proteins and peptides is complicated by the lack of activity assays that evaluate the properties of the whole molecule, as chem1 and chem2 both respond to the nonapeptide. This means that activity on those receptors is not indicative of the whole protein being in the correct conformation.
The solubility of peptides derived from C-terminus of chemerin may also be an issue. The sequence in chemerin between amino acids #148–156 (FYFPGQFAF) is hydrophobic, and peptides, including the nonapeptide, containing them may present solubility issues in aqueous solutions, thereby preventing accurate determinations of binding and activity parameters.
A 20-mer peptide whose C-terminus is identical to the nonapeptide has also been demonstrated to be active and can inhibit steroidogenesis in both rat testes and ovaries [31].
A defined structure of the nonapeptide has been proposed that consists of anti-parallel strands linked with a turn. This allows the formation of a hydrophobic core and may stabilize the amino acids that interact with the receptors. A model of the interaction of this peptide with chem1 has been developed based on mutational studies of chem1 and varying the composition of the peptide, showing that the critical binding is between F8 in the peptide (equivalent to residue 156F in chemerin) and a hydrophobic pocket in chem1, and F6 (equivalent to residue 154F in chemerin) with Y268 in chem1 [32]. The evidence for the model of the peptide was strengthened by a synthesis of cyclic peptides based on the model which retain activity. Those cyclic peptidomimetics can be modified to be stable in plasma for more than 48 h by modifying the N-terminus by substituting L-Tyr with D-Tyr [33].
The nonapeptide has also been used as the basis for designing an imaging agent for PET scanning by attaching a chelating agent via a linker to the N-terminus of the peptidomimetic [34]. In this case the peptidomimetic has been modified extensively with unnatural amino acids to prevent degradation while retaining activity. The resulting agent could be used to image DU4475 cancer cells on the flank of a mouse while not producing a clear image of A549 cells on the opposite flank [34]. The fact that DU4475 cells express chem1 while A549 cells do not suggests the specificity of the binding of the labeled peptidomimetic. The peptidomimetic without the chelator and linker has been used to demonstrate that it stimulates the growth of human colorectal cancer cells in a mouse xenograft model [35].
The key role of the C-terminal peptide that terminates at Ser157 has been confirmed in studies of the activity of different chemerin forms using the full-length protein on human chem1, which showed that chem157S was most active [27][36][27,36]. In contrast, chem158K and chem156F both possess about 5% of chem157S while the precursor, chem163S, has <5%, and chem155A is inactive. In a signal transduction assay, however, using COS cells transiently transfected with chem1, chem156F was found to be equipotent to chem157S [37]. Studies of the activity of different chemerin forms on chem2 have not been carried out in similar detail.
These studies require highly purified proteins because chem157S is so much more active than the other forms on chem1. If the sample of a purified form other than chem157S contains any trace amounts of chem157S, whether generated during production, purification, or in the assay, the activity of the sample will be overestimated. The same points also apply to the nonapeptide as they can also lead to overvaluing the activity of that form. In addition, it is important that the assay does not process the material being tested into an active form, so the reliability of any assay for this purpose is increased if the assay is fast, such as the measurement of calcium transients.
The sequence of the active nonapeptide is completely conserved between mouse and human, while there is overall high homology throughout the proteins (Figure 3a). It is notable, however, that the relative activities of mouse chem156S and chem155F differ from that of the equivalent human chem157S and chem 156F on either human or mouse chem1. Both mouse chem156S and human chem157S are equally active on chem1 from either species, but mouse chem155F is equipotent to mouse chem156S while human chem156F possesses only ~5% of the activity of human chem157S [11][38][9,38]. This suggests that in mice, chem155F may play a more important role as a ligand for chem1 than chem156F does in humans.
The nonapeptide has been used successfully in animal models to demonstrate differences from untreated animals in models such as abdominal aortic aneurysm and atherogenesis [39][40][39,40]. The data from the use of both the unmodified nonapeptide as well as the modified analogs support the concept that the key interactions are between F156 and/or S157 and chem1 and chem2.
Most papers reporting chemerin activity have been centered on chem1, although more recent reports have provided similar data sets for chem2. Chemerin is secreted as a precursor (chem163S) with low activity, but chem157S is fully active, and chem156F has partial activity. Chem163S does not spontaneously generate chem157S, which implies proteolytic processing of chem163S into its active forms and then their subsequent inactivation [7][26][27][7,26,27]. Many enzymes from the coagulation, fibrinolytic, and inflammatory systems have been identified that will process chem163S into active forms as well as inactivate those forms [7][41][42][7,41,42].
While the C-terminal tail of chemerin can be of different lengths, the N-terminal amino acid is E20 with no variation reported, as determined either by tryptic digest followed by MALDI-TOF or by Edman degradation [7][27][7,27]. This suggests that the N-terminus is relatively proteolytically stable.
A peptide (C15) was identified as being able to inhibit macrophage migration in a screen of 15–20 amino acid peptides based on high performance throughout assay. This peptide was from the C-terminus of chemerin containing residues A141-A155. It was very potent in inhibiting macrophage migration with a U-shaped dose response curve and maximum effect between 0.1–1 nM compared with full length chemerin with a maximum of 1–10 nM [43]. Treatment with this peptide, C15, was able to ameliorate the damage in a murine model of ischemic/reperfusion injury [44][45][44,45]. However, the inhibitory effects of chemerin on macrophages ex vivo could not be repeated by an independent group even though those macrophages express chem1 [46].
Completely separate to chemerin’s signaling via the processed C-terminal tail binding to its cognate receptors is its activity as an anti-bacterial defensin [47]. Unlike the interactions with chem1 and chem2, the anti-bacterial properties of chemerin do not depend on cleavage of the C-terminus. Instead, the activity is located within the disulfide bonded core of chemerin in the sequence from V66 to P85. This peptide alone is sufficient for strong inhibition of E. coli growth.