The matricellular protein osteopontin modulates cell–matrix interactions during tissue injury and healing. A complex multidomain structure of osteopontin enables it not only to bind diverse cell receptors but also to interact with various partners, including other extracellular matrix proteins, cytokines, and growth factors. Numerous studies have implicated osteopontin in the development and progression of myocardial remodeling in diverse cardiac diseases. Osteopontin influences myocardial remodeling by regulating extracellular matrix production, the activity of matrix metalloproteinases and various growth factors, inflammatory cell recruitment, myofibroblast differentiation, cardiomyocyte apoptosis, and myocardial vascularization. The exploitation of osteopontin loss- and gain-of-function approaches in rodent models provided an opportunity for assessment of the cell- and disease-specific contribution of osteopontin to myocardial remodeling.
- osteopontin
- right ventricle
- left ventricle
- heart failure
1. Introduction
2. Osteopontin in Heart Failure
Embryogenesis is associated with the augmented expression of osteopontin in various tissues and organs, including the heart [56,57,58][32][33][34]. Despite this implied function during embryogenesis, mice lacking osteopontin grow to maturity without any overt signs of morphological and functional cardiac abnormalities [29,59][29][35]. In line with this, in homeostatic conditions, osteopontin expression in the adult heart is very low [58[34][36][37][38][39],60,61,62,63], although it is regulated in a cell-specific manner. For example, a low basal expression of osteopontin was reported in cardiomyocytes [64][40], while it can be readily detected in coronary artery endothelial cells and cardiac fibroblasts [64,65][40][41]. Nevertheless, osteopontin-null mice respond to a variety of pathological stimuli, including tissue injury, inflammation, and infection, differently from wild-type mice [66][42]. In the last two decades, several studies utilizing osteopontin depletion either by neutralizing antibodies or by targeted mutagenesis have further advanced ouresearchers' understanding of the role of osteopontin in various cardiac pathologies [59,67,68,69,70,71,72][35][43][44][45][46][47][48]. Osteopontin-null mice were used in a number of HF models, including angiotensin-II (Ang-II) infusion [59][35], aldosterone infusion [67][43], transverse aortic constriction (TAC) [68][44], desmin-deficient model of dilated cardiomyopathy (DCM) [69][45], streptozotocin-induced model of diabetic cardiomyopathy [70][46], left anterior descending artery (LAD) ligation as a model of myocardial infarction (MI) [71][47] and a brief, repetitive LAD-occlusion model of ischemia-reperfusion (IR)-induced myocardial injury [72][48]. However, these and other studies have yielded conflicting results suggesting disease- and cell-specific roles of osteopontin in cardiac pathologies.2.1. Cell-Specific Regulation of Osteopontin
2.1.1. Osteopontin in Cardiomyocytes
Several factors have been shown to induce osteopontin expression in cardiomyocytes including aldosterone [73][49], dexamethasone [64][40], endothelin-1 [58][34], norepinephrine (NE) [58][34], and ROS [74][50] (Figure 21). In contrast, other known stimulators of osteopontin expression such as Ang-II [65[41][51],75], interleukin-1β (IL-1β), and interferon-γ (IFN-γ) [64][40] failed to induce osteopontin expression in cardiomyocytes.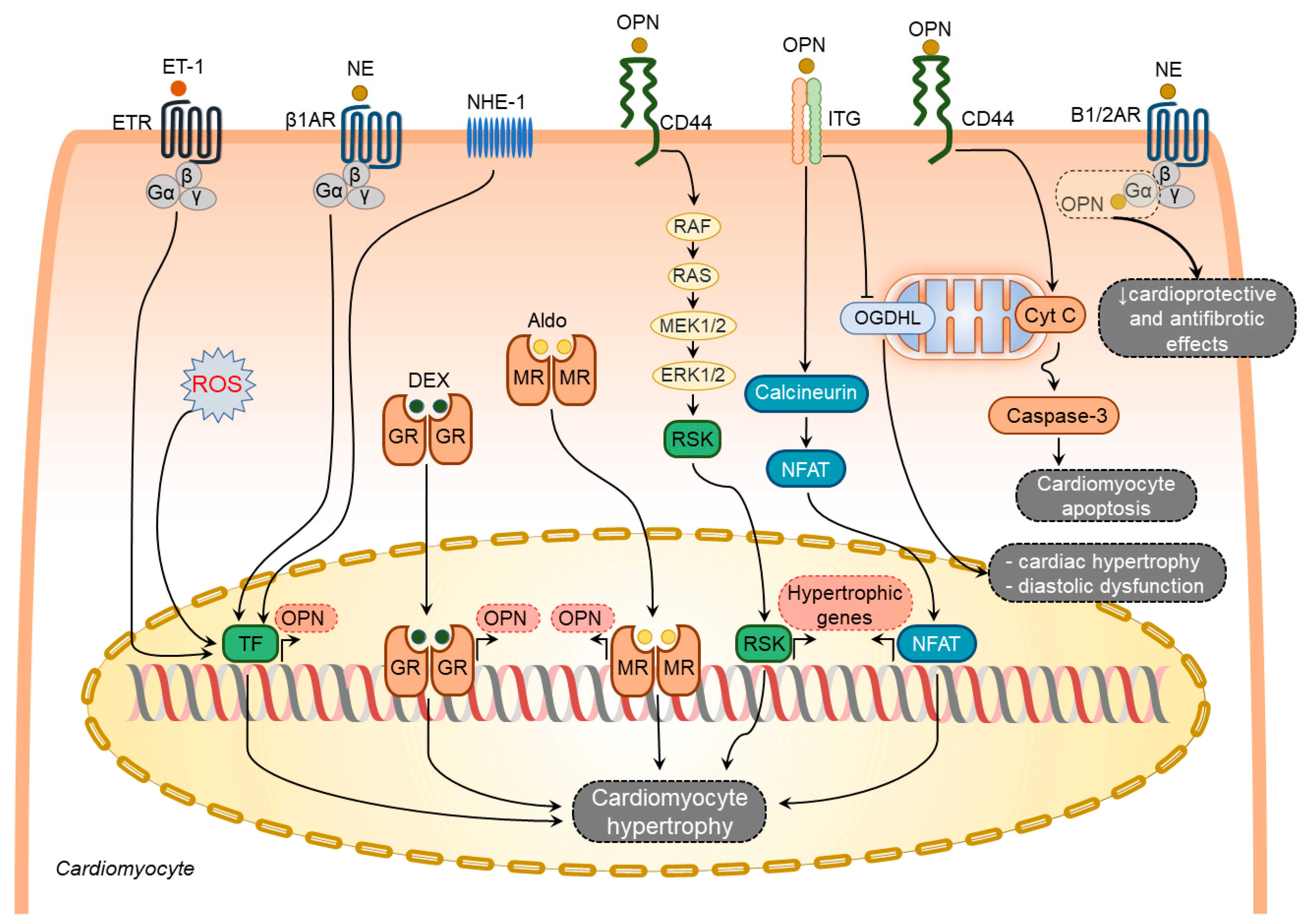
2.1.2. Osteopontin in Cardiac Fibroblasts
In DCM patients, increased levels of myocardial osteopontin strongly correlated with collagen expression [81][57], suggesting osteopontin involvement in cardiac fibrogenesis. Plasma from aged osteopontin-null mice failed to cause age-specific activation of cardiac fibroblasts [82][58]. Similarly, cardiac fibroblasts isolated from osteopontin-null mice displayed decreased proliferation and adhesion to ECM [59,83][35][59], less spreading, less resistance to detachment by shear stress, and a reduction in collagen gel contraction, which could be partially restored by ectopic osteopontin expression [83][59]. Several growth factors and cytokines have been shown to regulate osteopontin expression in cardiac fibroblasts, including Ang-II [65,75][41][51] and syndecan-4 [84][60] (Figure 32). Ang-II enhances osteopontin expression in cardiac fibroblasts via NADPH-ROS-mediated activation of ERK1/2 and JNKs pathways [65][41]. Syndecan-4 increases osteopontin expression via calcineurin/NFAT signaling pathways [84][60]. Mechanical stretch can also induce osteopontin expression in cardiac fibroblasts [85][61].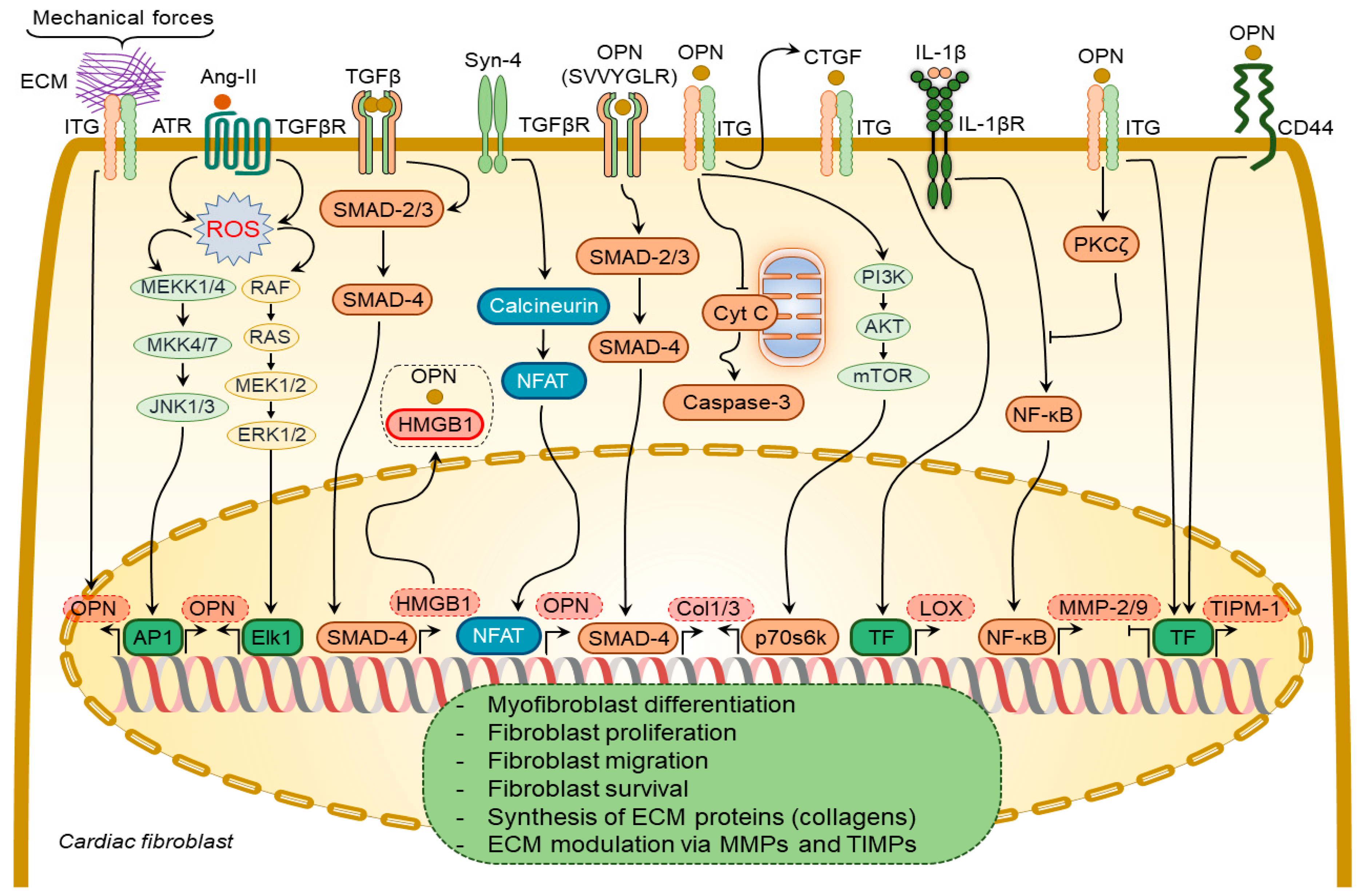
2.1.3. Osteopontin in Cardiac Endothelial Cells
Various growth factors and inflammatory mediators have been shown to regulate osteopontin expression in cardiac endothelial cells [64,65,75][40][41][51] (Figure 43). Ang-II increased osteopontin expression in cardiac endothelial cells [65,75][41][51] through NADPH-ROS-mediated activation of the Erk1/2 signaling pathway [75][51]. A combination of IL-1β and IFN-γ also augmented osteopontin expression in cardiac endothelial cells [64][40]. Moreover, dexamethasone significantly increased osteopontin expression in in vitro experiments [53][68].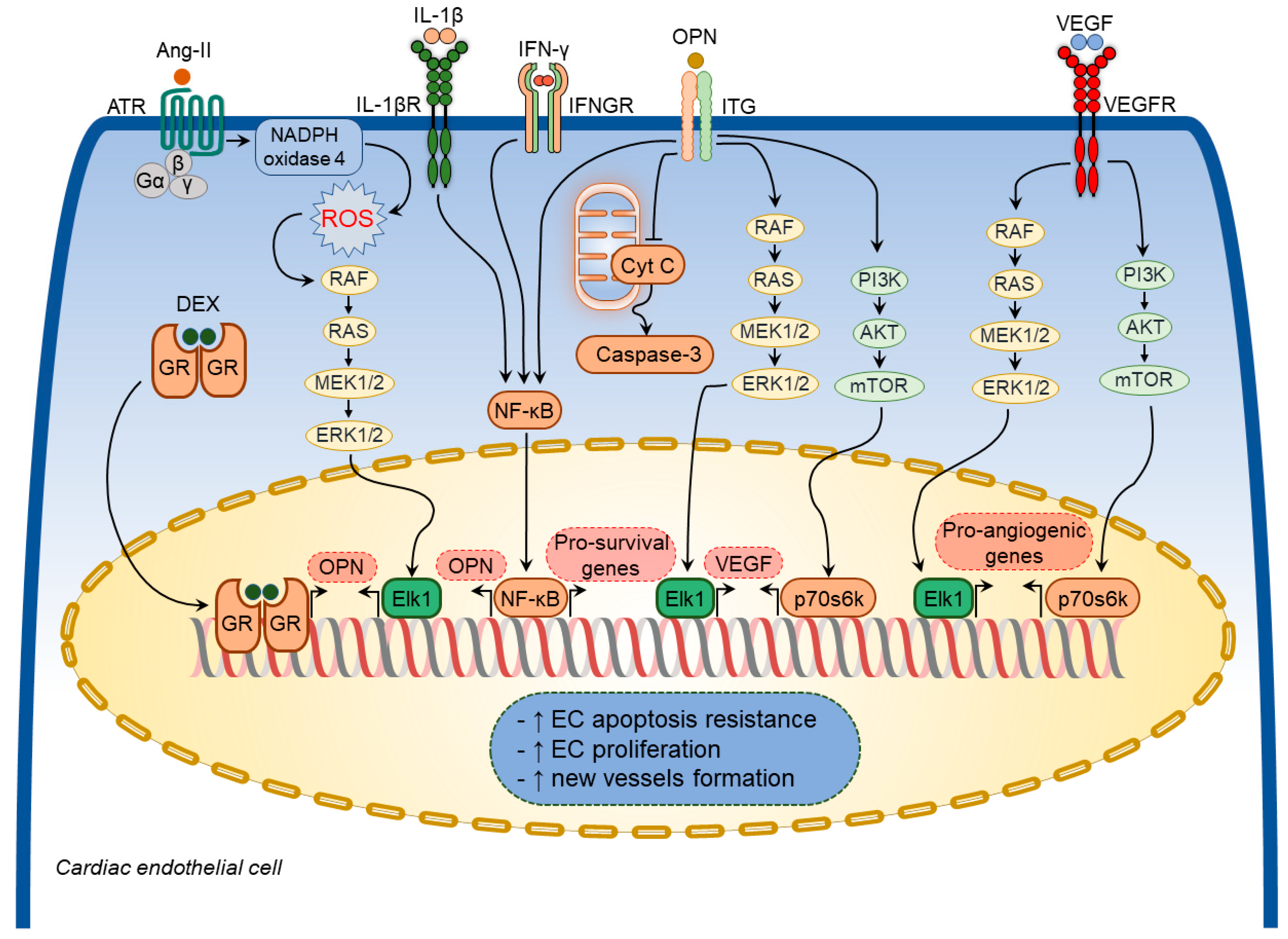
2.2. Implications of Osteopontin in Specific Pathological Processes
2.2.1. Osteopontin in Cardiac Hypertrophy
Cardiac hypertrophy is an adaptive response to hemodynamic load characterized by the thickening of the heart walls and enlargement of the individual cardiomyocytes, which has a compensatory role in maintaining cardiac performance and attenuating ventricular wall stress and chamber dilatation. At the cellular level, cardiomyocyte hypertrophy is characterized by an increase in cell dimensions mainly due to augmented contractile protein synthesis [101][74]. However, sustained hemodynamic stress in pathological conditions such as hypertension and valvular heart disease drives adverse alterations in the myocardium characterized by excessive cardiomyocyte apoptosis, myocardial inflammation, and fibrosis, eventually culminating in ventricular dilation and dysfunction. Robust upregulation of myocardial osteopontin expression upon mechanical stress was clearly shown in several rodent models [58,68,85][34][44][61]. Pressure overload induced an acute and strong elevation of osteopontin expression in the myocardium in rodents following TAC [58,68,85][34][44][61] and in rats with renovascular hypertension [58][34]. The increase in myocardial osteopontin expression was reported to be proportional to the degree of the afterload [102][75]. In line with these findings, there was a strong correlation between expression levels of osteopontin and atrial natriuretic peptide in the LV myocardium in renovascular and TAC-induced LV remodeling models [58][34]. In the latter models, cardiomyocytes were the main cell type in the heart producing osteopontin [58][34]. In contrast, in spontaneously hypertensive rats with cardiac hypertrophy, osteopontin expression was identified primarily in non-myocytes in the interstitial and perivascular space (possibly infiltrating macrophages and fibroblasts) [103][76]. The exploitation of osteopontin-null mice in several animal models of cardiac hypertrophy revealed that osteopontin is a key mediator in the mechanical stress-induced myocardial hypertrophic response [59,68,76,80][35][44][52][56]. In mice subjected to TAC, a lack of osteopontin was associated with attenuated cardiac hypertrophic response [68][44]. Knocking out osteopontin in transgenic mice with NHE1 overexpression significantly reduced cardiac hypertrophy, attenuated collagen deposition, and improved cardiac function [76][52]. Similarly, in another genetic mouse model of HF with preserved ejection fraction induced by Col4a3 deficiency, osteopontin deletion was associated with improved parameters of LV diastolic function and cardiac hypertrophy reduction [80][56]. Controversial data were obtained in the model of Ang-II infusion [29,59][29][35]. Thus, in one study, osteopontin deficiency attenuated cardiac hypertrophy induced by Ang-II infusion [59][35], whereas, in another study, osteopontin deletion did not prevent cardiac hypertrophy, despite attenuated myocardial fibrosis [29].2.2.2. Osteopontin in Cardiac Inflammation
Heart diseases are associated with inflammation, which contributes to cardiac dysfunction and myocardial fibrosis [10,104,105][10][77][78]. Numerous cytokines and chemokines are actively involved in the onset and progression of myocardial fibrosis [10,105][10][78]. Osteopontin has been shown to exert cytokine- and chemokine-like functions [35][79]. Among various inflammatory cells, macrophages represent the dominant inflammatory cell population in the remodeled myocardium [106][80]. Macrophage infiltration of the myocardium has been shown in various models, including spontaneously hypertensive rats and murine TAC [106,107][80][81]. Complete abolition of CD45+ monocytes recruitment to the myocardium in response to Ang-II infusion in osteopontin-null mice [108][82] suggested the involvement of osteopontin in this process. Osteopontin expressed in circulating leucocytes may also be actively involved in inflammatory cell recruitment to the myocardium and systemic inflammation in patients with various cardiac diseases [109][83]. Circulating CD4+ T lymphocytes expressing osteopontin and circulating osteopontin levels both correlate with the New York Heart Association Functional Class (NYHA FC) in HF patients and are associated with plaque instability in coronary artery disease patients [109,110][83][84]. Inflammatory cells have been identified as one of the major contributors to fibrogenesis in different tissues and organs under various disease conditions [111][85]. During the acute inflammatory phase of wound healing processes upon tissue injury in different tissues and organs, osteopontin expression is enhanced in infiltrating leukocytes, while during the chronic inflammatory phase, its upregulation is observed in resident macrophages [34][86]. Increased osteopontin expression by resident and recruited inflammatory cells increases collagen deposition and accumulation in the tissues [109,110,112][83][84][87]. Osteopontin expression by immune/inflammatory cells is associated with cardiac hypertrophic and fibrotic responses in the settings of a number of heart diseases [62,63,69,108,113][38][39][45][82][88]. Studies exploiting mice with a genetically modified osteopontin expression provided important insights into its role in myocardial inflammation in diverse cardiac conditions [69,79,112,114,115,116,117][45][55][87][89][90][91][92]. Mice overexpressing osteopontin in cardiomyocytes spontaneously develop severe cardiomyopathy, which is characterized by enhanced recruitment of inflammatory cells to the myocardium and excessive collagen accumulation, subsequently leading to chronic myocarditis and eventually premature death [79,112][55][87]. In line with these reports, deletion of osteopontin in desmin deficient mice, which spontaneously develop DCM with increased myocardial inflammation and fibrosis, was associated with an attenuation of myocardial inflammation and improvement in LV systolic function [69][45]. Cardiomyocyte-specific integrin-linked kinase (ILK) deficient mice spontaneously develop lethal cardiomyopathy characterized by excessive inflammatory cell accumulation, myocardial fibrosis, and cardiomyocyte apoptosis [115][90]. Augmented osteopontin expression in cardiomyocytes revealed by comprehensive profiling and mitigation of HF severity in these mice by application of anti-osteopontin antibodies implicated osteopontin as a major contributor to this phenotype [115][90]. Likewise, in other murine models, including those of Chagas heart disease and viral myocarditis, osteopontin-null mice displayed improved cardiac remodeling and attenuated myocardial inflammation [116][91] [117][92]. When taken together, osteopontin is highly upregulated in acute and chronic inflammatory phases of the myocardial injury and can contribute to adverse myocardial remodeling and cardiac dysfunction by promoting the recruitment of inflammatory cells to the myocardium.2.3. Clinical Implication of Osteopontin
Clinical studies have suggested that osteopontin might serve as a potent diagnostic and prognostic biomarker in diverse HF conditions. In this section, wresearchers discuss the roles and clinical implications of osteopontin in various HF diseases, including DCM, hypertensive HF, MI, and right HF (Figure 54).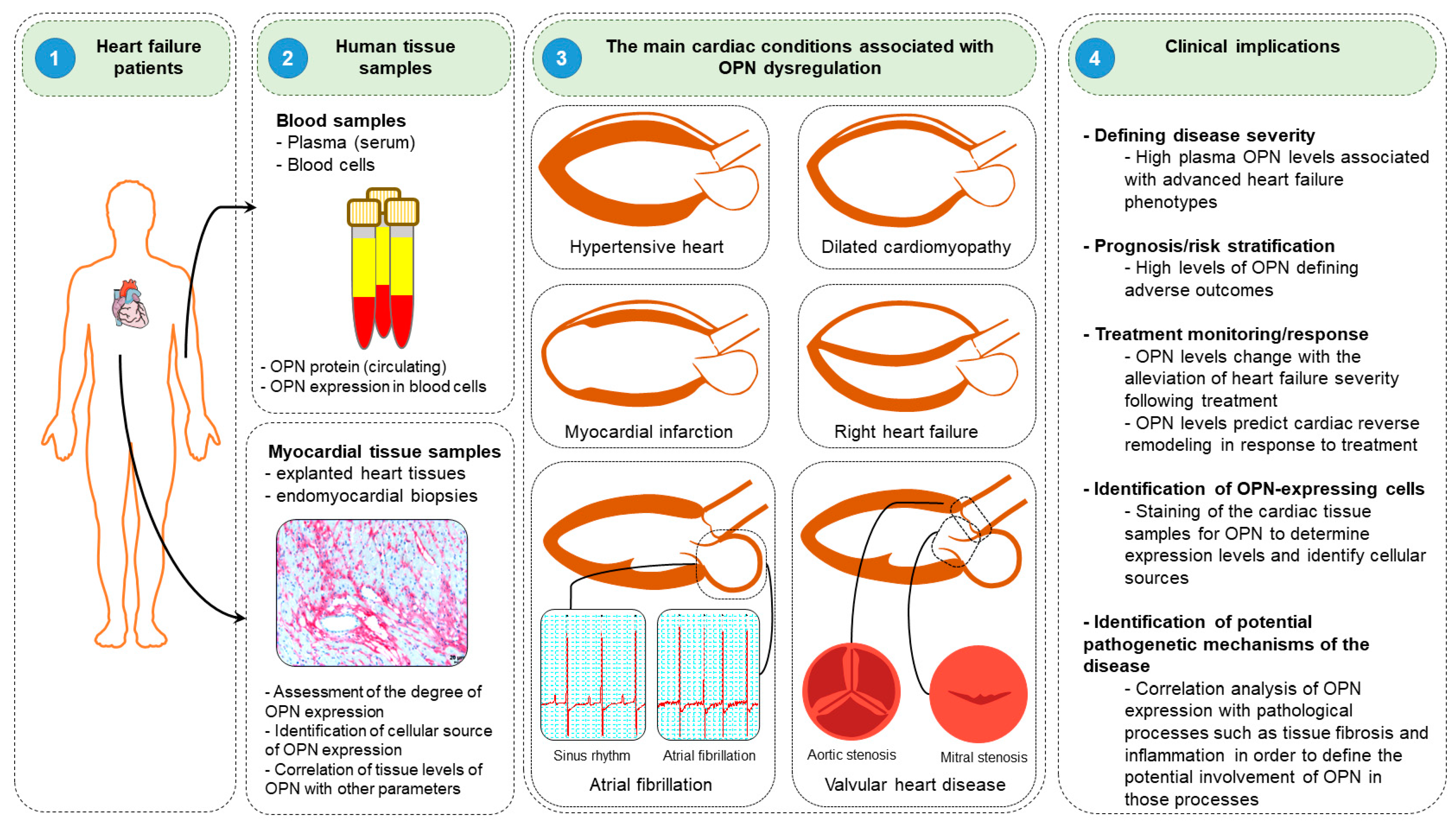
References
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; Amann, M.; Anderson, H.R.; Andrews, K.G.; Aryee, M.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2224–2260.
- Vos, T.; Abajobir, A.A.; Abate, K.H.; Abbafati, C.; Abbas, K.M.; Abd-Allah, F.; Abdulkader, R.S.; Abdulle, A.M.; Abebo, T.A.; Abera, S.F. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259.
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L. 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2013, 62, e147–e239.
- Corris, P.A.; Seeger, W. Call It by the Correct Name—Pulmonary Hypertension Not Pulmonary Arterial Hypertension: Growing Recognition of the Global Health Impact for a Well-Recognized Condition and the Role of the Pulmonary Vascular Research Institute; American Physiological Society Bethesda: Rockville, MD, USA, 2020.
- Friedberg, M.K.; Redington, A.N. Right versus left ventricular failure: Differences, similarities, and interactions. Circulation 2014, 129, 1033–1044.
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407.
- Vonk-Noordegraaf, A.; Haddad, F.; Chin, K.M.; Forfia, P.R.; Kawut, S.M.; Lumens, J.; Naeije, R.; Newman, J.; Oudiz, R.J.; Provencher, S.; et al. Right heart adaptation to pulmonary arterial hypertension: Physiology and pathobiology. J. Am. Coll. Cardiol. 2013, 62 (Suppl. S25), D22–D33.
- Egemnazarov, B.; Crnkovic, S.; Nagy, B.M.; Olschewski, H.; Kwapiszewska, G. Right ventricular fibrosis and dysfunction: Actual concepts and common misconceptions. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 68–69, 507–521.
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040.
- Sydykov, A.; Mamazhakypov, A.; Petrovic, A.; Kosanovic, D.; Sarybaev, A.S.; Weissmann, N.; Ghofrani, H.A.; Schermuly, R.T. Inflammatory Mediators Drive Adverse Right Ventricular Remodeling and Dysfunction and Serve as Potential Biomarkers. Front. Physiol. 2018, 9, 609.
- Carrillo-Salinas, F.J.; Ngwenyama, N.; Anastasiou, M.; Kaur, K.; Alcaide, P. Heart Inflammation: Immune Cell Roles and Roads to the Heart. Am. J. Pathol. 2019, 189, 1482–1494.
- Frump, A.L.; Bonnet, S.; de Jesus Perez, V.A.; Lahm, T. Emerging role of angiogenesis in adaptive and maladaptive right ventricular remodeling in pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L443–L460.
- Oka, T.; Akazawa, H.; Naito, A.T.; Komuro, I. Angiogenesis and cardiac hypertrophy: Maintenance of cardiac function and causative roles in heart failure. Circ. Res. 2014, 114, 565–571.
- Viswanathan, G.; Mamazhakypov, A.; Schermuly, R.T.; Rajagopal, S. The Role of G Protein-Coupled Receptors in the Right Ventricle in Pulmonary Hypertension. Front. Cardiovasc. Med. 2018, 5, 179.
- Hartupee, J.; Mann, D.L. Neurohormonal activation in heart failure with reduced ejection fraction. Nat. Rev. Cardiol. 2017, 14, 30–38.
- Ryan, J.J.; Archer, S.L. Emerging concepts in the molecular basis of pulmonary arterial hypertension: Part I: Metabolic plasticity and mitochondrial dynamics in the pulmonary circulation and right ventricle in pulmonary arterial hypertension. Circulation 2015, 131, 1691–1702.
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513.
- Piao, L.; Marsboom, G.; Archer, S.L. Mitochondrial metabolic adaptation in right ventricular hypertrophy and failure. J. Mol. Med. 2010, 88, 1011–1020.
- Sabbah, H.N. Targeting the Mitochondria in Heart Failure: A Translational Perspective. JACC Basic Transl. Sci. 2020, 5, 88–106.
- Shults, N.V.; Melnyk, O.; Suzuki, D.I.; Suzuki, Y.J. Redox Biology of Right-Sided Heart Failure. Antioxidants 2018, 7, 106.
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435.
- Esfandiary, A.; Kutsche, H.S.; Schreckenberg, R.; Weber, M.; Pak, O.; Kojonazarov, B.; Sydykov, A.; Hirschhauser, C.; Wolf, A.; Haag, D.; et al. Protection against pressure overload-induced right heart failure by uncoupling protein 2 silencing. Cardiovasc. Res. 2019, 115, 1217–1227.
- López, B.; Ravassa, S.; Moreno, M.U.; José, G.S.; Beaumont, J.; González, A.; Díez, J. Diffuse myocardial fibrosis: Mechanisms, diagnosis and therapeutic approaches. Nat. Rev. Cardiol. 2021, 18, 479–498.
- Frangogiannis, N.G. The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Investig. 2017, 127, 1600–1612.
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146.
- Frangogiannis, N.G. Matricellular proteins in cardiac adaptation and disease. Physiol. Rev. 2012, 92, 635–688.
- Singh, M.; Dalal, S.; Singh, K. Osteopontin: At the cross-roads of myocyte survival and myocardial function. Life Sci. 2014, 118, 1–6.
- Zahradka, P. Novel role for osteopontin in cardiac fibrosis. Circ. Res. 2008, 102, 270–272.
- Matsui, Y.; Jia, N.; Okamoto, H.; Kon, S.; Onozuka, H.; Akino, M.; Liu, L.; Morimoto, J.; Rittling, S.R.; Denhardt, D.; et al. Role of osteopontin in cardiac fibrosis and remodeling in angiotensin II-induced cardiac hypertrophy. Hypertension 2004, 43, 1195–1201.
- Anwar, A.; Li, M.; Frid, M.G.; Kumar, B.; Gerasimovskaya, E.V.; Riddle, S.R.; McKeon, B.A.; Thukaram, R.; Meyrick, B.O.; Fini, M.A.; et al. Osteopontin is an endogenous modulator of the constitutively activated phenotype of pulmonary adventitial fibroblasts in hypoxic pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L1–L11.
- Singh, M.; Foster, C.R.; Dalal, S.; Singh, K. Role of osteopontin in heart failure associated with aging. Heart Fail. Rev. 2010, 15, 487–494.
- Giachelli, C.M.; Schwartz, S.M.; Liaw, L. Molecular and cellular biology of osteopontin Potential role in cardiovascular disease. Trends Cardiovasc. Med. 1995, 5, 88–95.
- Thayer, J.M.; Giachelli, C.M.; Mirkes, P.E.; Schwartz, S.M. Expression of osteopontin in the head process late in gastrulation in the rat. J. Exp. Zool. 1995, 272, 240–244.
- Graf, K.; Do, Y.S.; Ashizawa, N.; Meehan, W.P.; Giachelli, C.M.; Marboe, C.C.; Fleck, E.; Hsueh, W.A. Myocardial osteopontin expression is associated with left ventricular hypertrophy. Circulation 1997, 96, 3063–3071.
- Collins, A.R.; Schnee, J.; Wang, W.; Kim, S.; Fishbein, M.C.; Bruemmer, D.; Law, R.E.; Nicholas, S.; Ross, R.S.; Hsueh, W.A. Osteopontin modulates angiotensin II-induced fibrosis in the intact murine heart. J. Am. Coll. Cardiol. 2004, 43, 1698–1705.
- Singh, M.; Foster, C.R.; Dalal, S.; Singh, K. Osteopontin: Role in extracellular matrix deposition and myocardial remodeling post-MI. J. Mol. Cell. Cardiol. 2010, 48, 538–543.
- Denhardt, D.T.; Guo, X. Osteopontin: A protein with diverse functions. FASEB J. 1993, 7, 1475–1482.
- Williams, E.B.; Halpert, I.; Wickline, S.; Davison, G.; Parks, W.C.; Rottman, J.N. Osteopontin expression is increased in the heritable cardiomyopathy of Syrian hamsters. Circulation 1995, 92, 705–709.
- Murry, C.E.; Giachelli, C.M.; Schwartz, S.M.; Vracko, R. Macrophages express osteopontin during repair of myocardial necrosis. Am. J. Pathol. 1994, 145, 1450–1462.
- Singh, K.; Balligand, J.L.; Fischer, T.A.; Smith, T.W.; Kelly, R.A. Glucocorticoids increase osteopontin expression in cardiac myocytes and microvascular endothelial cells. Role in regulation of inducible nitric oxide synthase. J. Biol. Chem. 1995, 270, 28471–28478.
- Xie, Z.; Singh, M.; Singh, K. ERK1/2 and JNKs, but not p38 kinase, are involved in reactive oxygen species-mediated induction of osteopontin gene expression by angiotensin II and interleukin-1beta in adult rat cardiac fibroblasts. J. Cell Physiol. 2004, 198, 399–407.
- Lund, S.A.; Giachelli, C.M.; Scatena, M. The role of osteopontin in inflammatory processes. J. Cell Commun. Signal. 2009, 3, 311–322.
- Sam, F.; Xie, Z.; Ooi, H.; Kerstetter, D.L.; Colucci, W.S.; Singh, M.; Singh, K. Mice lacking osteopontin exhibit increased left ventricular dilation and reduced fibrosis after aldosterone infusion. Am. J. Hypertens. 2004, 17, 188–193.
- Xie, Z.; Singh, M.; Singh, K. Osteopontin modulates myocardial hypertrophy in response to chronic pressure overload in mice. Hypertension 2004, 44, 826–831.
- Psarras, S.; Mavroidis, M.; Sanoudou, D.; Davos, C.H.; Xanthou, G.; Varela, A.E.; Panoutsakopoulou, V.; Capetanaki, Y. Regulation of adverse remodelling by osteopontin in a genetic heart failure model. Eur. Heart J. 2012, 33, 1954–1963.
- Subramanian, V.; Krishnamurthy, P.; Singh, K.; Singh, M. Lack of osteopontin improves cardiac function in streptozotocin-induced diabetic mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H673–H683.
- Trueblood, N.A.; Xie, Z.; Communal, C.; Sam, F.; Ngoy, S.; Liaw, L.; Jenkins, A.W.; Wang, J.; Sawyer, D.B.; Bing, O.H.; et al. Exaggerated left ventricular dilation and reduced collagen deposition after myocardial infarction in mice lacking osteopontin. Circ. Res. 2001, 88, 1080–1087.
- Duerr, G.D.; Mesenholl, B.; Heinemann, J.C.; Zoerlein, M.; Huebener, P.; Schneider, P.; Feisst, A.; Ghanem, A.; Tiemann, K.; Dewald, D.; et al. Cardioprotective effects of osteopontin-1 during development of murine ischemic cardiomyopathy. BioMed Res. Int. 2014, 2014, 124063.
- Pollard, C.M.; Desimine, V.L.; Wertz, S.L.; Perez, A.; Parker, B.M.; Maning, J.; McCrink, K.A.; Shehadeh, L.A.; Lymperopoulos, A. Deletion of Osteopontin Enhances beta(2)-Adrenergic Receptor-Dependent Anti-Fibrotic Signaling in Cardiomyocytes. Int. J. Mol. Sci. 2019, 20, 1396.
- Jiang, P.; Zhang, D.; Qiu, H.; Yi, X.; Zhang, Y.; Cao, Y.; Zhao, B.; Xia, Z.; Wang, C. Tiron ameliorates high glucose-induced cardiac myocyte apoptosis by PKCdelta-dependent inhibition of osteopontin. Clin. Exp. Pharmacol. Physiol. 2017, 44, 760–770.
- Xie, Z.; Pimental, D.R.; Lohan, S.; Vasertriger, A.; Pligavko, C.; Colucci, W.S.; Singh, K. Regulation of angiotensin II-stimulated osteopontin expression in cardiac microvascular endothelial cells: Role of p42/44 mitogen-activated protein kinase and reactive oxygen species. J. Cell Physiol. 2001, 188, 132–138.
- Abdulrahman, N.; Jaspard-Vinassa, B.; Fliegel, L.; Jabeen, A.; Riaz, S.; Gadeau, A.P.; Mraiche, F. Na(+)/H(+) exchanger isoform 1-induced osteopontin expression facilitates cardiac hypertrophy through p90 ribosomal S6 kinase. Physiol. Genom. 2018, 50, 332–342.
- Mohamed, I.A.; Gadeau, A.P.; Fliegel, L.; Lopaschuk, G.; Mlih, M.; Abdulrahman, N.; Fillmore, N.; Mraiche, F. Na+/H+ exchanger isoform 1-induced osteopontin expression facilitates cardiomyocyte hypertrophy. PLoS ONE 2015, 10, e0123318.
- Mlih, M.; Abdulrahman, N.; Gadeau, A.P.; Mohamed, I.A.; Jaballah, M.; Mraiche, F. Na(+)/H (+) exchanger isoform 1 induced osteopontin expression in cardiomyocytes involves NFAT3/Gata4. Mol. Cell. Biochem. 2015, 404, 211–220.
- Dalal, S.; Zha, Q.; Daniels, C.R.; Steagall, R.J.; Joyner, W.L.; Gadeau, A.P.; Singh, M.; Singh, K. Osteopontin stimulates apoptosis in adult cardiac myocytes via the involvement of CD44 receptors, mitochondrial death pathway, and endoplasmic reticulum stress. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1182–H1191.
- Yousefi, K.; Irion, C.I.; Takeuchi, L.M.; Ding, W.; Lambert, G.; Eisenberg, T.; Sukkar, S.; Granzier, H.L.; Methawasin, M.; Lee, D.I.; et al. Osteopontin Promotes Left Ventricular Diastolic Dysfunction Through a Mitochondrial Pathway. J. Am. Coll. Cardiol. 2019, 73, 2705–2718.
- Cabiati, M.; Svezia, B.; Matteucci, M.; Botta, L.; Pucci, A.; Rinaldi, M.; Caselli, C.; Lionetti, V.; Del Ry, S. Myocardial Expression Analysis of Osteopontin and Its Splice Variants in Patients Affected by End-Stage Idiopathic or Ischemic Dilated Cardiomyopathy. PLoS ONE 2016, 11, e0160110.
- Sawaki, D.; Czibik, G.; Pini, M.; Ternacle, J.; Suffee, N.; Mercedes, R.; Marcelin, G.; Surenaud, M.; Marcos, E.; Gual, P.; et al. Visceral Adipose Tissue Drives Cardiac Aging Through Modulation of Fibroblast Senescence by Osteopontin Production. Circulation 2018, 138, 809–822.
- Lenga, Y.; Koh, A.; Perera, A.S.; McCulloch, C.A.; Sodek, J.; Zohar, R. Osteopontin expression is required for myofibroblast differentiation. Circ. Res. 2008, 102, 319–327.
- Herum, K.M.; Lunde, I.G.; Skrbic, B.; Louch, W.E.; Hasic, A.; Boye, S.; Unger, A.; Brorson, S.H.; Sjaastad, I.; Tonnessen, T.; et al. Syndecan-4 is a key determinant of collagen cross-linking and passive myocardial stiffness in the pressure-overloaded heart. Cardiovasc. Res. 2015, 106, 217–226.
- Herum, K.M.; Romaine, A.; Wang, A.; Melleby, A.O.; Strand, M.E.; Pacheco, J.; Braathen, B.; Duner, P.; Tonnessen, T.; Lunde, I.G.; et al. Syndecan-4 Protects the Heart From the Profibrotic Effects of Thrombin-Cleaved Osteopontin. J. Am. Heart Assoc. 2020, 9, e013518.
- Zhao, H.; Wang, W.; Zhang, J.; Liang, T.; Fan, G.P.; Wang, Z.W.; Zhang, P.D.; Wang, X.; Zhang, J. Inhibition of osteopontin reduce the cardiac myofibrosis in dilated cardiomyopathy via focal adhesion kinase mediated signaling pathway. Am. J. Transl. Res. 2016, 8, 3645–3655.
- Ashizawa, N.; Graf, K.; Do, Y.S.; Nunohiro, T.; Giachelli, C.M.; Meehan, W.P.; Tuan, T.L.; Hsueh, W.A. Osteopontin is produced by rat cardiac fibroblasts and mediates A(II)-induced DNA synthesis and collagen gel contraction. J. Clin. Investig. 1996, 98, 2218–2227.
- Xie, Z.; Singh, M.; Siwik, D.A.; Joyner, W.L.; Singh, K. Osteopontin inhibits interleukin-1beta-stimulated increases in matrix metalloproteinase activity in adult rat cardiac fibroblasts: Role of protein kinase C-zeta. J. Biol. Chem. 2003, 278, 48546–48552.
- Pardo, A.; Gibson, K.; Cisneros, J.; Richards, T.J.; Yang, Y.; Becerril, C.; Yousem, S.; Herrera, I.; Ruiz, V.; Selman, M.; et al. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2005, 2, e251.
- Lindsey, M.L.; Zouein, F.A.; Tian, Y.; Padmanabhan Iyer, R.; de Castro Bras, L.E. Osteopontin is proteolytically processed by matrix metalloproteinase 9. Can. J. Physiol. Pharmacol. 2015, 93, 879–886.
- Lopez, B.; Gonzalez, A.; Lindner, D.; Westermann, D.; Ravassa, S.; Beaumont, J.; Gallego, I.; Zudaire, A.; Brugnolaro, C.; Querejeta, R.; et al. Osteopontin-mediated myocardial fibrosis in heart failure: A role for lysyl oxidase? Cardiovasc. Res. 2013, 99, 111–120.
- Kim, J.S.; Bashir, M.M.; Werth, V.P. Gottron’s papules exhibit dermal accumulation of CD44 variant 7 (CD44v7) and its binding partner osteopontin: A unique molecular signature. J. Investig. Dermatol. 2012, 132, 1825–1832.
- Dai, J.; Peng, L.; Fan, K.; Wang, H.; Wei, R.; Ji, G.; Cai, J.; Lu, B.; Li, B.; Zhang, D.; et al. Osteopontin induces angiogenesis through activation of PI3K/AKT and ERK1/2 in endothelial cells. Oncogene 2009, 28, 3412–3422.
- Khan, S.A.; Lopez-Chua, C.A.; Zhang, J.; Fisher, L.W.; Sorensen, E.S.; Denhardt, D.T. Soluble osteopontin inhibits apoptosis of adherent endothelial cells deprived of growth factors. J. Cell. Biochem. 2002, 85, 728–736.
- Scatena, M.; Almeida, M.; Chaisson, M.L.; Fausto, N.; Nicosia, R.F.; Giachelli, C.M. NF-kappaB mediates alphavbeta3 integrin-induced endothelial cell survival. J. Cell Biol. 1998, 141, 1083–1093.
- Liaw, L.; Lindner, V.; Schwartz, S.M.; Chambers, A.F.; Giachelli, C.M. Osteopontin and beta 3 integrin are coordinately expressed in regenerating endothelium in vivo and stimulate Arg-Gly-Asp-dependent endothelial migration in vitro. Circ. Res. 1995, 77, 665–672.
- Zhao, X.; Johnson, J.N.; Singh, K.; Singh, M. Impairment of myocardial angiogenic response in the absence of osteopontin. Microcirculation 2007, 14, 233–240.
- Frey, N.; Katus, H.A.; Olson, E.N.; Hill, J.A. Hypertrophy of the heart: A new therapeutic target? Circulation 2004, 109, 1580–1589.
- Ndisang, J.F.; Chibbar, R.; Lane, N. Heme oxygenase suppresses markers of heart failure and ameliorates cardiomyopathy in L-NAME-induced hypertension. Eur. J. Pharmacol. 2014, 734, 23–34.
- Singh, K.; Sirokman, G.; Communal, C.; Robinson, K.G.; Conrad, C.H.; Brooks, W.W.; Bing, O.H.; Colucci, W.S. Myocardial osteopontin expression coincides with the development of heart failure. Hypertension 1999, 33, 663–670.
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112.
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the role of inflammation in heart failure. Nat. Rev. Cardiol. 2020, 17, 269–285.
- Icer, M.A.; Gezmen-Karadag, M. The multiple functions and mechanisms of osteopontin. Clin. Biochem. 2018, 59, 17–24.
- Weisheit, C.; Zhang, Y.; Faron, A.; Köpke, O.; Weisheit, G.; Steinsträsser, A.; Frede, S.; Meyer, R.; Boehm, O.; Hoeft, A.; et al. Ly6C(low) and not Ly6C(high) macrophages accumulate first in the heart in a model of murine pressure-overload. PLoS ONE 2014, 9, e112710.
- Yang, G.H.; Zhou, X.; Ji, W.J.; Zeng, S.; Dong, Y.; Tian, L.; Bi, Y.; Guo, Z.Z.; Gao, F.; Chen, H.; et al. Overexpression of VEGF-C attenuates chronic high salt intake-induced left ventricular maladaptive remodeling in spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H598–H609.
- Lorenzen, J.M.; Schauerte, C.; Hubner, A.; Kolling, M.; Martino, F.; Scherf, K.; Batkai, S.; Zimmer, K.; Foinquinos, A.; Kaucsar, T.; et al. Osteopontin is indispensible for AP1-mediated angiotensin II-related miR-21 transcription during cardiac fibrosis. Eur. Heart J. 2015, 36, 2184–2196.
- Soejima, H.; Irie, A.; Fukunaga, T.; Oe, Y.; Kojima, S.; Kaikita, K.; Kawano, H.; Sugiyama, S.; Yoshimura, M.; Kishikawa, H.; et al. Osteopontin expression of circulating T cells and plasma osteopontin levels are increased in relation to severity of heart failure. Circ. J. Off. J. Jpn. Circ. Soc. 2007, 71, 1879–1884.
- Soejima, H.; Irie, A.; Fukunaga, T.; Sugamura, K.; Kojima, S.; Sakamoto, T.; Yoshimura, M.; Kishikawa, H.; Nishimura, Y.; Ogawa, H. Elevated plasma osteopontin levels were associated with osteopontin expression of CD4+ T cells in patients with unstable angina. Circ. J. Off. J. Jpn. Circ. Soc. 2006, 70, 851–856.
- Van Linthout, S.; Miteva, K.; Tschöpe, C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc. Res. 2014, 102, 258–269.
- Mazzali, M.; Kipari, T.; Ophascharoensuk, V.; Wesson, J.; Johnson, R.; Hughes, J. Osteopontin—A molecule for all seasons. Qjm 2002, 95, 3–13.
- Renault, M.A.; Robbesyn, F.; Reant, P.; Douin, V.; Daret, D.; Allieres, C.; Belloc, I.; Couffinhal, T.; Arnal, J.F.; Klingel, K.; et al. Osteopontin expression in cardiomyocytes induces dilated cardiomyopathy. Circ. Heart Fail. 2010, 3, 431–439.
- Satoh, M.; Nakamura, M.; Akatsu, T.; Shimoda, Y.; Segawa, I.; Hiramori, K. Myocardial osteopontin expression is associated with collagen fibrillogenesis in human dilated cardiomyopathy. Eur. J. Heart Fail. 2005, 7, 755–762.
- Papathanasiou, S.; Rickelt, S.; Soriano, M.E.; Schips, T.G.; Maier, H.J.; Davos, C.H.; Varela, A.; Kaklamanis, L.; Mann, D.L.; Capetanaki, Y. Tumor necrosis factor-alpha confers cardioprotection through ectopic expression of keratins K8 and K18. Nat. Med. 2015, 21, 1076–1084.
- Dai, J.; Matsui, T.; Abel, E.D.; Dedhar, S.; Gerszten, R.E.; Seidman, C.E.; Seidman, J.G.; Rosenzweig, A. Deep sequence analysis of gene expression identifies osteopontin as a downstream effector of integrin-linked kinase (ILK) in cardiac-specific ILK knockout mice. Circ. Heart Fail. 2014, 7, 184–193.
- Caballero, E.P.; Santamaria, M.H.; Corral, R.S. Endogenous osteopontin induces myocardial CCL5 and MMP-2 activation that contributes to inflammation and cardiac remodeling in a mouse model of chronic Chagas heart disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 11–23.
- Szalay, G.; Sauter, M.; Haberland, M.; Zuegel, U.; Steinmeyer, A.; Kandolf, R.; Klingel, K. Osteopontin: A fibrosis-related marker molecule in cardiac remodeling of enterovirus myocarditis in the susceptible host. Circ. Res. 2009, 104, 851–859.