Pulmonary fibrosis (PF) is a feared outcome of many pulmonary diseases which results in a reduction in lung compliance and capacity. The development of PF is relatively rare, but it can occur secondary to viral pneumonia, especially COVID-19 infection. While COVID-19 infection and its complications are still under investigation, we can look at a similar outbreak in the past to gain better insight as to the expected long-term outcomes of COVID-19 patient lung function.
Pulmonary fibrosis (PF) is a feared outcome of many pulmonary diseases which results in a reduction in lung compliance and capacity. The development of PF is relatively rare, but it can occur secondary to viral pneumonia, especially COVID-19 infection. While COVID-19 infection and its complications are still under investigation, researchers can look at a similar outbreak in the past to gain better insight as to the expected long-term outcomes of COVID-19 patient lung function.
- pulmonary fibrosis
- COVID-19
- TGF-β1
- prevention
1. Introduction
2. Pathophysiology of Pulmonary Fibrosis
The pathophysiology of PF is an area of ongoing research. This disease state can result from many etiologies which may result in different primary mechanisms that drive the shared fibrotic outcome. Patients who develop acute respiratory distress syndrome (ARDS) [5] may experience in an excessive proliferation of fibrotic factors leading to pulmonary fibrosis. Other patients may develop fibrosis from environmental insults, such as chest irradiation or chronic occupational exposure to irritants such as asbestos or silica. Rarely, patients may develop IPF, which is a severe, progressive fibrotic disease of pulmonary tissue with an unknown insult (Figure 1).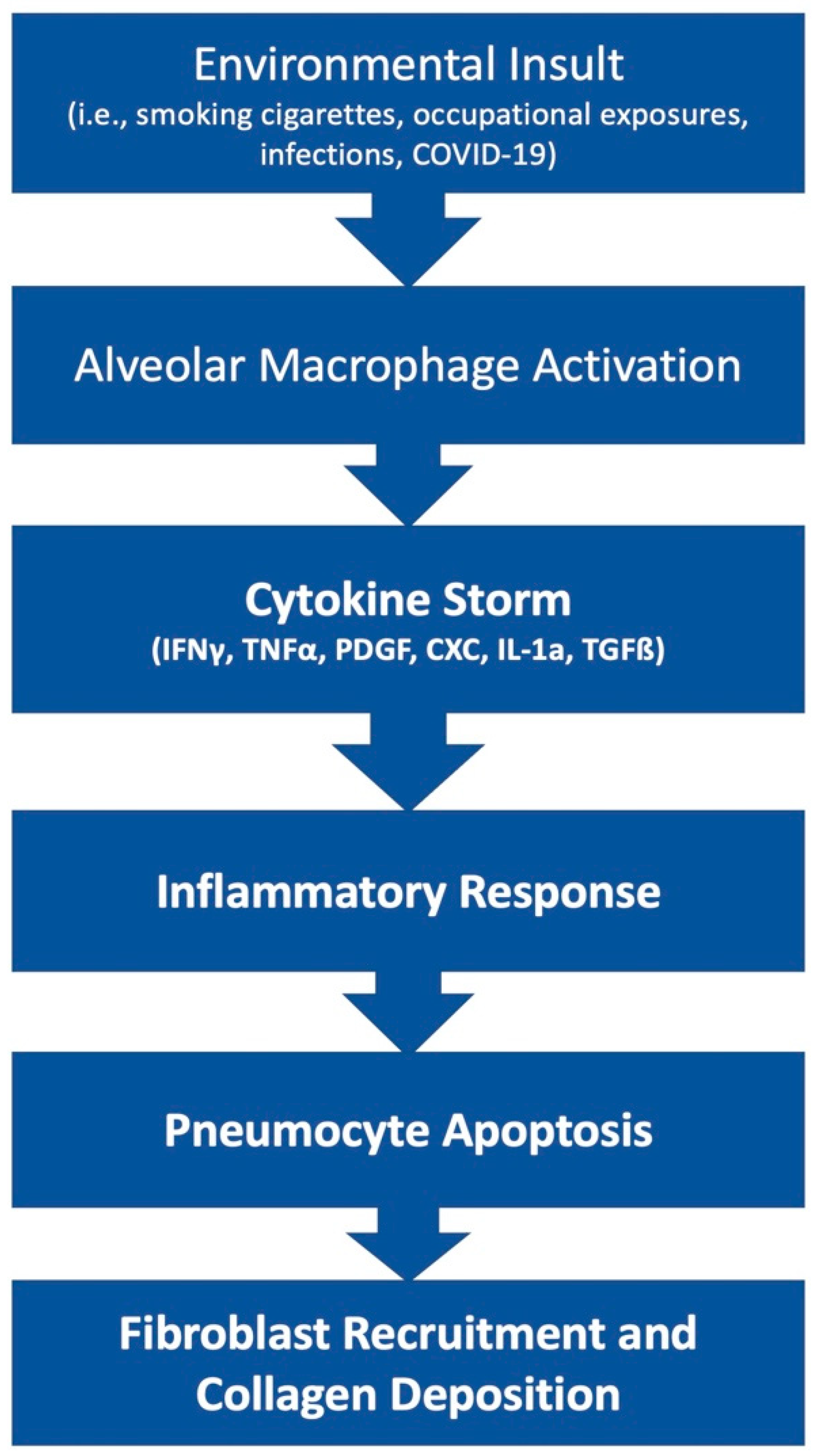
References
- Gentile, F.; Aimo, A.; Forfori, F.; Catapano, G.; Clemente, A.; Cademartiri, F.; Emdin, M.; Giannoni, A. COVID-19 and risk of pulmonary fibrosis: The importance of planning ahead. Eur. J. Prev. Cardiol. 2020, 27, 1442–1446.
- Hui, D.S.; Wong, K.T.; Ko, F.W.; Tam, L.S.; Chan, D.P.; Woo, J.; Sung, J.J. The 1-year impact of severe acute respiratory syndrome on pulmonary function, exercise capacity, and quality of life in a cohort of survivors. Chest 2005, 128, 2247–2261.
- Ngai, J.C.; Ko, F.W.; Ng, S.S.; To, K.W.; Tong, M.; Hui, D.S. The long-term impact of severe acute respiratory syndrome on pulmonary function, exercise capacity and health status. Respirology 2010, 15, 543–550.
- Chang, Y.C.; Yu, C.J.; Chang, S.C.; Galvin, J.R.; Liu, H.M.; Hsiao, C.H.; Kuo, P.H.; Chen, K.Y.; Franks, T.J.; Huang, K.M.; et al. Pulmonary sequelae in convalescent patients after severe acute respiratory syndrome: Evaluation with thin-section CT. Radiology 2005, 236, 1067–1075.
- Tomashefski, J.F., Jr. Pulmonary pathology of acute respiratory distress syndrome. Clin. Chest Med. 2000, 21, 435–466.
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar epithelial type II cells as drivers of lung fibrosis in idiopathic pulmonary fibrosis. Int. J. Mol. Sci. 2020, 21, 2269.
- Katzen, J.; Beers, M.F. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J. Clin. Investig. 2020, 130, 5088–5099.
- Yue, X.; Shan, B.; Lasky, J.A. TGF-β: Titan of Lung Fibrogenesis. Curr. Enzym. Inhib. 2010, 6, 67–77.
- Zhang, Y.; Jiao, H.; Wu, Y.; Sun, X. P120-catenin regulates pulmonary fibrosis and TGF-β induced lung fibroblast differentiation. Life Sci. 2019, 230, 35–44.
- Chanda, D.; Otoupalova, E.; Smith, S.R.; Volckaert, T.; De Langhe, S.P.; Thannickal, V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Asp. Med. 2019, 65, 56–69.
- Xiao, L.; Du, Y.; Shen, Y.; He, Y.; Zhao, H.; Li, Z. TGF-beta 1 induced fibroblast proliferation is mediated by the FGF-2/ERK pathway. Front. Biosci. 2012, 17, 2667–2674.
- Oda, K.; Yatera, K.; Izumi, H.; Ishimoto, H.; Yamada, S.; Nakao, H.; Hanaka, T.; Ogoshi, T.; Noguchi, S.; Mukae, H. Profibrotic role of WNT10A via TGF-ß signaling in idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 39.
- Kikuchi, N.; Ishii, Y.; Morishima, Y.; Yageta, Y.; Haraguchi, N.; Itoh, K.; Yamamoto, M.; Hizawa, N. Nrf2 protects against pulmonary fibrosis by regulating the lung oxidant level and Th1/Th2 balance. Respir. Res. 2010, 11, 31.
- Kolahian, S.; Fernandez, I.E.; Eickelberg, O.; Hartl, D. Immune Mechanisms in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 55, 309–322.
- Agostini, C.; Gurrieri, C. Chemokine/cytokine cocktail in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 357–363.
- Upagupta, C.; Shimbori, C.; Alsilmi, R.; Kolb, M. Matrix abnormalities in pulmonary fibrosis. Eur. Respir. Rev. 2018, 27, 180033.
- Wei, P.; Xie, Y.; Abel, P.W.; Huang, Y.; Ma, Q.; Li, L.; Hao, J.; Wolff, D.W.; Wei, T.; Tu, Y. Transforming growth factor (TGF)-β1-induced miR-133a inhibits myofibroblast differentiation and pulmonary fibrosis. Cell Death Dis. 2019, 10, 670.
- Li, H.; Zhao, C.; Tian, Y.; Lu, J.; Zhang, G.; Liang, S.; Chen, D.; Liu, X.; Kuang, W.; Zhu, M. Src family kinases and pulmonary fibrosis: A review. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 127, 110183.
- Cheresh, P.; Kim, S.J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1028–1040.
- Suh, Y.J.; Hong, H.; Ohana, M.; Bompard, F.; Revel, M.P.; Valle, C.; Gervaise, A.; Poissy, J.; Susen, S.; Hékimian, G.; et al. Pulmonary Embolism and Deep Vein Thrombosis in COVID-19: A Systematic Review and Meta-Analysis. Radiology 2021, 298, E70–E80.