Centipedes are typical venomous arthropods that rely on their toxins primarily for predation and defense. Venoms are a complex cocktail of biologically active molecules, including peptides, proteins, polyamide, and enzymes widely produced by venomous organisms. Through long-term evolution, venomous animals have evolved highly specific and diversified peptides and proteins targeting key physiological elements, including the nervous system.
- venom
- centipede
- peptide
- protein
1. Introduction
Centipedes are excellent predatory arthropods. They deploy a broad set of bioactive peptides to capture prey or defend against predators [23,38,47,48,49,50][1][2][3][4][5][6]. Neurotoxins are the primary predation and defense peptides in centipede venom and also important ingredients that have made significant progress in revealing the biological activities and action mechanisms in recent research. These components act on a wide array of targets, mostly the ion channels, either by activating or inhibiting their electric activity.
2. Toxins Targeting Voltage-Gated Sodium Channels
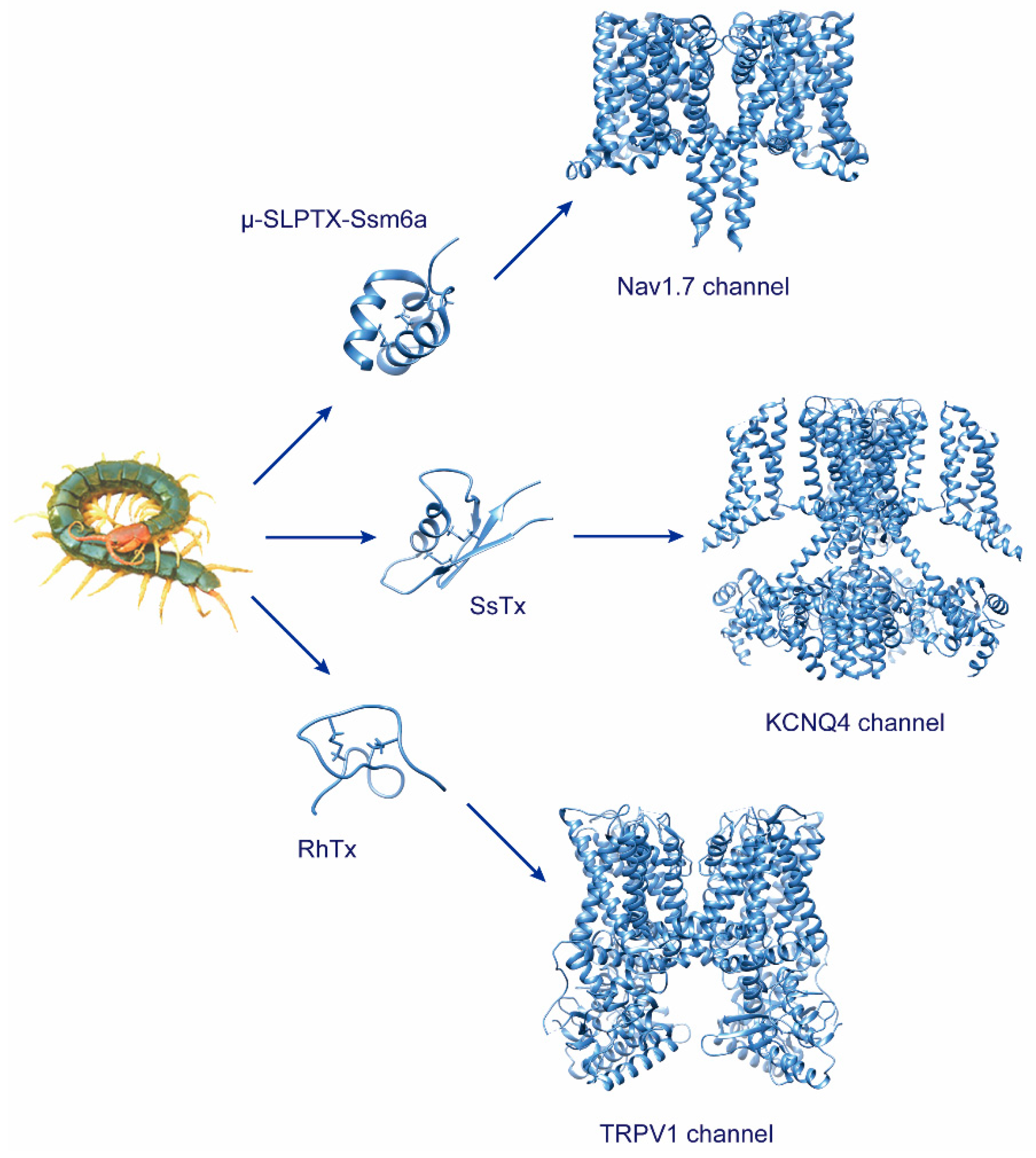
3. Toxins Targeting Voltage-Gated Potassium Channels
4. Toxins Targeting Voltage-Gated Calcium Channels
5. Toxins Targeting TRPV1 Channel
References
- Tang, D.; Xu, J.; Li, Y.; Zhao, P.; Kong, X.; Hu, H.; Liang, S.; Tang, C.; Liu, Z. Molecular mechanisms of centipede toxin SsTx-4 inhibition of inwardly rectifying potassium channels. J. Biol. Chem. 2021, 297, 101076.
- Undheim, E.A.; Fry, B.G.; King, G.F. Centipede venom: Recent discoveries and current state of knowledge. Toxins 2015, 7, 679–704.
- Zhao, F.; Lan, X.; Li, T.; Xiang, Y.; Zhao, F.; Zhang, Y.; Lee, W.H. Proteotranscriptomic Analysis and Discovery of the Profile and Diversity of Toxin-like Proteins in Centipede. Mol. Cell. Proteom. 2018, 17, 709–720.
- Chu, Y.; Qiu, P.; Yu, R. Centipede Venom Peptides Acting on Ion Channels. Toxins 2020, 12, 230.
- Dash, T.S.; Shafee, T.; Harvey, P.J.; Zhang, C.; Peigneur, S.; Deuis, J.R.; Vetter, I.; Tytgat, J.; Anderson, M.A.; Craik, D.J.; et al. A Centipede Toxin Family Defines an Ancient Class of CSalphabeta Defensins. Structure 2019, 27, 315–326.e7.
- Hakim, M.A.; Yang, S.; Lai, R. Centipede venoms and their components: Resources for potential therapeutic applications. Toxins 2015, 7, 4832–4851.
- Nguyen, P.T.; Yarov-Yarovoy, V. Towards Structure-Guided Development of Pain Therapeutics Targeting Voltage-Gated Sodium Channels. Front. Pharmacol. 2022, 13, 842032.
- Payandeh, J.; Scheuer, T.; Zheng, N.; Catterall, W.A. The crystal structure of a voltage-gated sodium channel. Nature 2011, 475, 353–358.
- Yang, S.; Liu, Z.; Xiao, Y.; Li, Y.; Rong, M.; Liang, S.; Zhang, Z.; Yu, H.; King, G.F.; Lai, R. Chemical punch packed in venoms makes centipedes excellent predators. Mol. Cell. Proteom. 2012, 11, 640–650.
- Yang, S.; Xiao, Y.; Kang, D.; Liu, J.; Li, Y.; Undheim, E.A.; Klint, J.K.; Rong, M.; Lai, R.; King, G.F. Discovery of a selective NaV1.7 inhibitor from centipede venom with analgesic efficacy exceeding morphine in rodent pain models. Proc. Natl. Acad. Sci. USA 2013, 110, 17534–17539.
- Xiao, Y.; Blumenthal, K.; Jackson, J.O., 2nd; Liang, S.; Cummins, T.R. The tarantula toxins ProTx-II and huwentoxin-IV differentially interact with human Nav1.7 voltage sensors to inhibit channel activation and inactivation. Mol. Pharmacol. 2010, 78, 1124–1134.
- Wright, Z.V.F.; McCarthy, S.; Dickman, R.; Reyes, F.E.; Sanchez-Martinez, S.; Cryar, A.; Kilford, I.; Hall, A.; Takle, A.K.; Topf, M.; et al. The Role of Disulfide Bond Replacements in Analogues of the Tarantula Toxin ProTx-II and Their Effects on Inhibition of the Voltage-Gated Sodium Ion Channel Nav1.7. J. Am. Chem. Soc. 2017, 139, 13063–13075.
- Liu, Z.C.; Zhang, R.; Zhao, F.; Chen, Z.M.; Liu, H.W.; Wang, Y.J.; Jiang, P.; Zhang, Y.; Wu, Y.; Ding, J.P.; et al. Venomic and transcriptomic analysis of centipede Scolopendra subspinipes dehaani. J. Proteome Res. 2012, 11, 6197–6212.
- González-Morales, L.; Pedraza-Escalona, M.; Diego-Garcia, E.; Restano-Cassulini, R.; Batista, C.V.; Gutiérrez Mdel, C.; Possani, L.D. Proteomic characterization of the venom and transcriptomic analysis of the venomous gland from the Mexican centipede Scolopendra viridis. J. Proteom. 2014, 111, 224–237.
- Rong, M.; Yang, S.; Wen, B.; Mo, G.; Kang, D.; Liu, J.; Lin, Z.; Jiang, W.; Li, B.; Du, C.; et al. Peptidomics combined with cDNA library unravel the diversity of centipede venom. J. Proteom. 2015, 114, 28–37.
- Luo, L.; Li, B.; Wang, S.; Wu, F.; Wang, X.; Liang, P.; Ombati, R.; Chen, J.; Lu, X.; Cui, J.; et al. Centipedes subdue giant prey by blocking KCNQ channels. Proc. Natl. Acad. Sci. USA 2018, 115, 1646–1651.
- Du, C.; Li, J.; Shao, Z.; Mwangi, J.; Xu, R.; Tian, H.; Mo, G.; Lai, R.; Yang, S. Centipede KCNQ Inhibitor SsTx Also Targets K(V)1.3. Toxins 2019, 11, 76.
- Ramu, Y.; Lu, Z. A family of orthologous proteins from centipede venoms inhibit the hKir6.2 channel. Sci. Rep. 2019, 9, 14088.
- Aguilar, M.B.; Perez-Reyes, L.I.; Lopez, Z.; de la Cotera, E.P.; Falcon, A.; Ayala, C.; Galvan, M.; Salvador, C.; Escobar, L.I. Peptide sr11a from Conus spurius is a novel peptide blocker for Kv1 potassium channels. Peptides 2010, 31, 1287–1291.
- Julius, D. TRP channels and pain. Annu. Rev. Cell Dev. Biol. 2013, 29, 355–384.
- Yang, S.; Yang, F.; Wei, N.; Hong, J.; Li, B.; Luo, L.; Rong, M.; Yarov-Yarovoy, V.; Zheng, J.; Wang, K.; et al. A pain-inducing centipede toxin targets the heat activation machinery of nociceptor TRPV1. Nat. Commun. 2015, 6, 8297.
- Luo, L.; Wang, Y.; Li, B.; Xu, L.; Kamau, P.M.; Zheng, J.; Yang, F.; Yang, S.; Lai, R. Molecular basis for heat desensitization of TRPV1 ion channels. Nat. Commun. 2019, 10, 2134.
- Zhu, A.; Aierken, A.; Yao, Z.; Vu, S.; Tian, Y.; Zheng, J.; Yang, S.; Yang, F. A centipede toxin causes rapid desensitization of nociceptor TRPV1 ion channel. Toxicon 2020, 178, 41–49.