Age-related macular degeneration (AMD) is the leading cause of irreversible vision loss in the elderly population. AMD is characterized in its late form by neovascularization (wet type) or geographic atrophy of the retinal pigment epithelium cell layer (dry type). Regarding the latter type, there is growing evidence supporting an association between the pathophysiology of dry AMD and key proteins in the complement cascade. The complement cascade works as a central part of the innate immune system by defending against foreign pathogens and modified self-tissues. Through three distinct pathways, a series of plasma and membrane-associated serum proteins are activated upon identification of a foreign entity. Several of these proteins have been implicated in the development and progression of dry AMD. Potential therapeutic targets include C1q, C3, C5, complement factors (B, D, H, I), membrane attack complex, and properdin.
- age-related macular degeneration
- AMD
- complement cascade
1. Introduction
2. General Pathogenesis of Dry AMD
Dry AMD is a multifactorial disease entity with various genetic and environmental elements contributing toward pathogenesis that remains entirely elusive. One component of this process is the overproduction of reactive oxygen species, which translates to oxidative stress, free radical formation, and peroxidation of the RPE [12][13][14][34,35,36]. Oxidative stress particularly obstructs the functionality of RPE stem cells by inhibiting mitochondrial activity [13][15][16][17][35,37,38,39], thereby creating a pro-inflammatory microenvironment unconducive to appropriate retinal development. Indeed, human RPE cells exposed to greater concentrations of oxidative stress demonstrate a dose-dependent reduction in viability [18][40]. Compounded with other factors, these oxidative insults result in the formation of drusen, which are foundational to the progression of dry AMD [19][41]. This formation and enlargement of drusen inhibits the ability of Bruch’s membrane (BM) to transport oxygen, nutrients, and metabolic substances by creating RPE detachments [20][42]. In turn, photoreceptor debris accumulates, resulting in further accretion of metabolites toxic to the RPE, such as lipofuscin and A2E [21][22][43,44]. Drusen additionally contain multiple pro-inflammatory markers, thus suggesting the importance of these processes towards the development of dry AMD [23][45]. These include components of the complement cascade, such as activators, fragments, and the membrane attack complex (MAC) [24][46]. The collective consequence of the aforementioned events is GA, a condition demarcated by outer retinal atrophic lesions that occur secondary to the loss or attenuation of the choriocapillaris, photoreceptors, and RPE [25][26][47,48].3. Complement Cascade
The complement cascade is a component of the innate and adaptive immune systems, possessing a critical role in the defense against pathogens and the maintenance of homeostasis. Three principal pathways are involved in complement: the classical, alternative, and lectin pathways, as highlighted in Figure 1. Each is comprised of a series of reactions that ultimately conclude in the creation of a MAC [27][49]; nonetheless, the proteins involved in the initiation of each mechanism are distinct.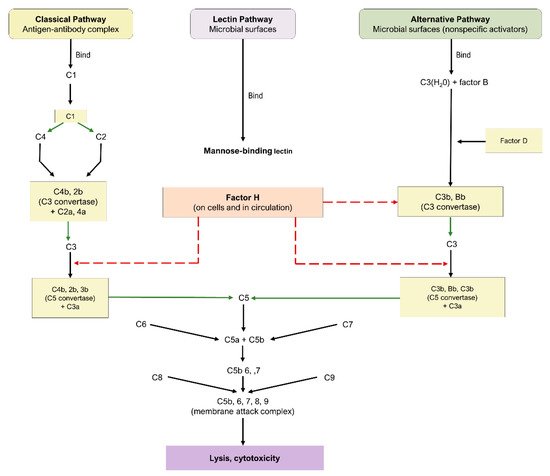
3.1. Pathways of the Complement Cascade
The classical pathway is initiated through the binding of antigen–antibody complexes to the C1q protein, the alternative pathway is initiated through a binding interaction between the C3b protein and foreign substances, and the lectin pathway involves the interaction of mannose-binding lectin with mannose-based polysaccharides on microbials. While the lectin and alternative pathways are effector arms of innate immunity, the classical pathway is an effector arm of adaptive immunity. Nevertheless, all three pathways converge at C3 convertase, leading to the cleavage of C3 into C3a and c3b, formation of C5 convertase, cleavage of C5 into C5a and c5b, and eventual formation of the MAC and subsequent cell destruction [27][49]. As a consequence of their fundamental roles, C3, C5, and the MAC have emerged as crucial targets for the therapeutic pipeline of dry AMD.3.2. Complement Cascade in AMD
As alluded to previously, overactivity of the complement pathway has been heavily implicated in the pathogenesis of drusen [28][50]. Several proteins involved in the complement system have been identified as possible actors in the development and progression of dry AMD, including C3, C5, CFH, CFI, complement factor D (CFD), and complement factor B (CFB). Evidence of this association is abundant in both experimental and clinical investigations. Among mice with inactivated C3, the rate of CNV was significantly lower following laser photocoagulation, thereby highlighting the protein’s role in the pathogenesis of AMD [29][30][51,52]. Other investigations have reported the presence of C5 in drusen and its elevation in the bloodstream of patients with AMD. C5a has been identified as having numerous inflammatory actions and thus is an effective target in reducing retinal loss in murine models [31][32][53,54]. MAC tends to accumulate in regions adjacent to microvascular injury, an early indicator of AMD; therefore, modulating MAC may represent a mechanism to prevent and/or limit the progression of GA [31][33][34][53,55,56]. Furthermore, downstream products of CFB have been observed to be elevated in AMD patients, inactivation of CFD has been linked to decreased photoreceptor loss, CFP stabilizes C3 and C5 convertases, and CD59 has been a basis for gene therapy specific to MAC [35][36][37][57,58,59].4. Current Therapeutic Targets
ANX007 (Annexon Inc., Brisbane, CA, USA) is a recombinant monoclonal antibody with an antigen-binding fragment that inhibits c1q. Through its action on c1q, the classical complement pathway is inhibited alongside C3 and C5 [38][39][60,61]. In a murine model of retinal photooxidative damage, ANX-M1, a monoclonal anti-C1q antibody similar to ANX007, reduced retinal atrophy [40][62]. Furthermore, a phase I trial (NCT04188015) using 2.5 mg and 5 mg of intravitreal (IVT) ANX007 demonstrated the safety and tolerability of both doses in 17 patients with primary open-angle glaucoma [38][39][60,61].
AMY-106, derived from Cp40-KKK, a fourth-generation compstatin analog, is a novel C3 inhibitor in development by Amyndas Pharmaceuticals [41][42][63,64]. This therapeutic maintained intraocular residence for more than 90 days after one 0.5 mg IVT injection in an investigation involving cynomolgus monkeys. Moreover, AMY-106 exhibited notable retinal tissue penetrance, as it localized with C3 in the choriocapillaris [41][63]. Provided its promise for dry AMD treatment, a phase I trial of AMY-106 is in development [42][64].
Pegcetacoplan, additionally denoted as APL-2, is a PEGylated peptide inhibitor of C3 formulated by Apellis Pharmaceuticals that is administered intravitreally [43][65]. The agent was evaluated in the FILLY trial (NCT02503332) [43][65], a multicenter, randomized phase II trial encompassing 246 patients with GA. Subjects were randomized in a 2:2:1:1 ratio to injections of 15 mg pegcetacoplan monthly or EOM, or sham injections monthly or EOM for 12 months. Follow-up was conducted at 15 and 18 months. Pegcetacoplan treatment produced statistically significant reductions in GA growth rates.
POT-4 (AL-78898A) is a compstatin analog originally developed by Potentia Pharmaceuticals that functions as a C3 inhibitor and was the first complement inhibitor tested in humans with AMD [38][44][60,70]. Phase I trial (NCT00473928) results revealed no adverse drug events (ADEs) or serious adverse events (SAEs) with doses of up to 450 mg for patients with wet AMD [44][70]. Based on the tolerability of POT-4, a multicenter, randomized phase II trial (NCT01603043) with 10 patients was conducted to assess its efficacy in reducing GA lesion growth among individuals with dry AMD. However, the study was terminated prematurely, as four of seven participants (57.14%) in the POT-4 group developed drug product deposits in the eye.
NGM621 (NGM Biopharmaceuticals, San Francisco, CA, USA) is a humanized immunoglobulin G1 monoclonal antibody formulated as a C3 inhibitor. A phase I trial (NCT04014777) included 15 patients with GA secondary to dry AMD [45][71]. Participants were treated with either single-ascending doses (2 mg, 7.5 mg, 15 mg) of NGM621 or two doses of 15 mg delivered 4 weeks apart. Monitoring occurred for 12 weeks within all cohorts, and results indicated an appropriate safety profile, with no SAEs, ADEs, or new-onset CNV reported.
Eculizumab (Alexion Pharmaceuticals, Inc) is a humanized monoclonal antibody directed against C5 [46][47][72,73]. Systemic eculizumab is currently approved for the treatment of paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Thus, considering the evidence for the relationship of the complement cascade with AMD, the COMPLETE study (NCT00935883) was designed [46][47][72,73].
Avacincaptad pegol (Zimura®), by Iveric Bio (Cranbury, NJ, USA), is a PEGylated RNA aptamer that operates as a C5 cleavage inhibitor, thereby impeding the complement cascade irrespective of the initial activation pathway. This agent was evaluated in the phase II/III GATHER1 trial (NCT02686658), a multicenter, randomized investigation. Randomization was conducted in two stages. In total, 77 part I participants were randomized in a 1:1:1 ratio to injections of 1 mg of avacincaptad pegol, 2 mg of avacincaptad pegol, and sham. Then, 209 part II participants were randomized in a 1:2:2 ratio to 2 mg of avacincaptad pegol, 4 mg of avacincaptad pegol, and sham. Relative to sham, subjects receiving 2 mg of avacincaptad pegol and 4 mg of avacincaptad pegol experienced a 27.4% (p = 0.0072) and 27.8% (p = 0.0051) reduction, respectively, in the mean rate of GA growth [48][74].
Tesidolumab (LFG316), a monoclonal C5 inhibitor developed by Novartis Pharmaceuticals (Basel, Switzerland), was investigated in a multicenter, randomized phase II clinical trial (NCT01527500). In the study, 158 patients were enrolled and separated into two groups. Part A examined the safety and efficacy of multiple 5 mg IVT injections of LFG316 relative to sham every 28 days over the course of 505 days. Part B examined the safety and pharmacokinetic properties of a single 10 mg IVT injection. Results revealed a relatively benign safety profile, with no demonstrable improvement in the primary outcome of GA lesion growth or the secondary outcome of BCVA.
IONIS-FB-lrx (Ionis Pharmaceuticals, Carlsbad, CA, USA) is a novel anti-sense oligonucleotide (ASO) targeting the gene encoding complement factor B (CFB), a moiety of the alternative complement pathway. When delivered subcutaneously, IONIS-FB-lrx reduced circulating levels of CFB in a dose-dependent manner among 54 healthy participants of a phase I trial [49][76]. No notable adverse effects were reported. Given these results, the multicenter, randomized phase II GOLDEN trial (NCT03815825) has been initiated to evaluate the influence of IONIS-FB-lrx administration on the progression of GA lesion size [49][76].
Complement factor D (CFD), a rate-limiting enzyme of the alternative pathway, converts proconvertases into active C3 and C5 convertases. Consequently, inhibition of CFD has surfaced as an attractive therapeutic option for dry AMD [50][77]. Lampalizumab, designed by Genentech (San Francisco, CA, USA), is a humanized monoclonal antibody with an antigen-binding fragment that inhibits complement factor D. In a phase I trial (NCT00973011), 18 participants received 10 mg of IVT lampalizumab. The agent had an acceptable safety profile without notable ocular or systemic ADEs or SAEs. To expand these findings, the MAHALO phase II trial (NCT01229215) was conducted to evaluate lampalizumab’s efficacy in reducing GA progression [51][78]. Monthly treatment with lampalizumab resulted in a statistically significant 20% reduction (p = 0.117) in mean GA lesion enlargement relative to sham. Patients with high-risk complement factor I (CFI) alleles displayed a greater response to monthly lampalizumab with a 44% reduction (p = 0.0037) in GA atrophy progression compared to sham. Similar to phase I findings, lampalizumab was well-tolerated [51][78]. CFH downregulates the alternative complement cascade by preventing C3 convertase formation from c3b; therefore, it has been postulated as a potential therapeutic for dry AMD. The injection of AdCAGfH, an adeno-associated virus (AAV) containing murine factor H, demonstrated an attenuation of C3-induced retinal inflammation [52][81]. This gene therapy is currently under consideration for human subjects [53][82]. GEM103 is a recombinant human CFH developed by Gemini Therapeutics (Cambridge, MA, USA). In a phase I clinical trial (NCT04246866) comprised of 12 participants, GEM103 demonstrated minimal adverse effects and a dose-dependent increase in levels of CFH [54][83]. Significantly, no CNV was observed, a phenomenon previously reported in the literature in association with repeated administration of IVT complement inhibitors [55][67]. CFI is a regulator of the alternative pathway. With the assistance of its cofactors, it cleaves C3b into pro-inflammatory ic3b, which is subsequently cleaved into inert c3dg. Thus, the protein is crucial for modulating the activation of the alternative pathway. Importantly, experimental investigations confirmed the ability of AAV-containing CFI cDNA to induce CFI protein expression in human RPE lines and murine retina, thereby stimulating the development of GT005 by Gyroscope Therapeutics. Regulators of the complement cascade are emerging targets for investigational compounds. AAVCAGsCD59, also denoted HMR59, is an intravitreally injected AAV vector developed by Hemera Biosciences that induces the formation of CD59, thereby preventing the binding of C9 required for the formation of a complete MAC [56][88]. Pre-clinically, HMR59 demonstrated efficacy in attenuating laser-induced CNV among murine models [57][89]. Properdin is a positive regulator of the alternative complement pathway, preventing the degradation of C3 and C5 convertases. Thus, CLG561, an anti-properdin antibody, was developed as a potential therapeutic by Alcon Research [58][91]. Results from a phase I trial (NCT01835015) showed IVT doses of CLG561 up to 10 mg were safe and well-tolerated, with no systemic or ocular ADEs reported [59][92].