Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Sirius Huang and Version 2 by Sirius Huang.
The body is equipped with a “food factor-sensing system” that senses food factors, such as polyphenols, sulfur-containing compounds, and vitamins, taken into the body, and plays an essential role in manifesting their physiological effects. For example, (−)-epigallocatechin-3-O-gallate (EGCG), the representative catechin in green tea (Camellia sinensi L.), exerts various effects, including anti-cancer, anti-inflammatory, and anti-allergic effects, when sensed by the cell surficial protein 67-kDa laminin receptor (67LR).
- green tea
- catechin
- EGCG
- 67LR
- EGCG-sensing system
- anti-cancer
- anti-inflammation
- anti-allergy
1. Identification of the Green Tea Polyphenol EGCG-Sensing Receptor
The representative components responsible for the bioactivity of green tea are catechins, a subgroup of polyphenols. These catechins include EGCG, (−)-epigallocatechin (EGC), (−)-epicatechin-3-gallate (ECG), and (−)-epicatechin (EC). Their C-2 epimeric isomers are (−)-catechin (C), (−)-gallocatechin (GC), (−)-catechin-3-O-gallate (CG), and (−)-gallocatechin-3-O-gallate (GCG). The non-epicatechins (GCG, CG, GC, and C) in tea leaves are known to be less abundant than the epicatechins (EGCG, ECG, EGC, and EC). These four epicatechins are readily epimerized by heat treatment and pH conditions to produce the corresponding non-epimeric forms [1]. EGCG is found only in tea leaves from Camellia sinensis, and is therefore a characteristic component of green tea. It is the most abundant of the catechins and shows the strongest bioactivity among catechins. Pharmacokinetic studies in human subjects have indicated that the maximum plasma concentration (Cmax) after a dose of EGCG in our daily life is usually <1.0 μM. On the other hand, oral administration of an EGCG supplement or a standardized green tea extract Polyphenon ETM, containing 60% EGCG, has also shown that the plasma level of EGCG can reach as high as 7 μM [2]. Most pharmacological effects of EGCG observed in vitro experiment and cell-free systems have been obtained at concentrations (10–100 μM) considerably higher than those reported in vivo after intake of green tea or EGCG. Furthermore, the intracellular levels of EGCG were much lower than the extracellular ones. Identifying the proteins that EGCG exhibits high affinity towards is the first step toward deciphering the molecular mechanisms underlying its anti-cancer effect. Several such proteins have been identified using in vitro techniques [3][4]. All of them are essential to the anti-cancer effect of EGCG in cancer cells. However, for this effect to be observed, EGCG concentrations higher than the dissociation constant (Kd) values are required.
All-trans-retinoic acid (ATRA) amplifies the attachment of EGCG to the cancer cell surface. This molecular interaction was evaluated by using a surface plasmon resonance binding analysis. cDNA libraries were obtained from cancer cells either treated with ATRA or were untreated. Subtractive cloning was used to identify the differences in the composition of mRNA transcripts of the two types of cells. Thus, 67LR was identified as an isolated single target for EGCG and found to be essential to the EGCG binding to the cancer cell surface and, thus, to its inhibitory effect on cell growth at the concentration of 5 μM [5]. The EGCG binding site is located on the extracellular domain of 67LR, corresponding to the 161–170 region [6] (Figure 1). The calculated Kd value for the EGCG−67LR complex is about 40 nM. Most 67LR proteins are located in the region of lipid rafts, plasma membrane microdomains enriched in cholesterol and sphingolipids, rather than the non-lipid rafts [7]. This type of distribution matches the plasma membrane-associated EGCG concentration determined after treating cells with EGCG [8].
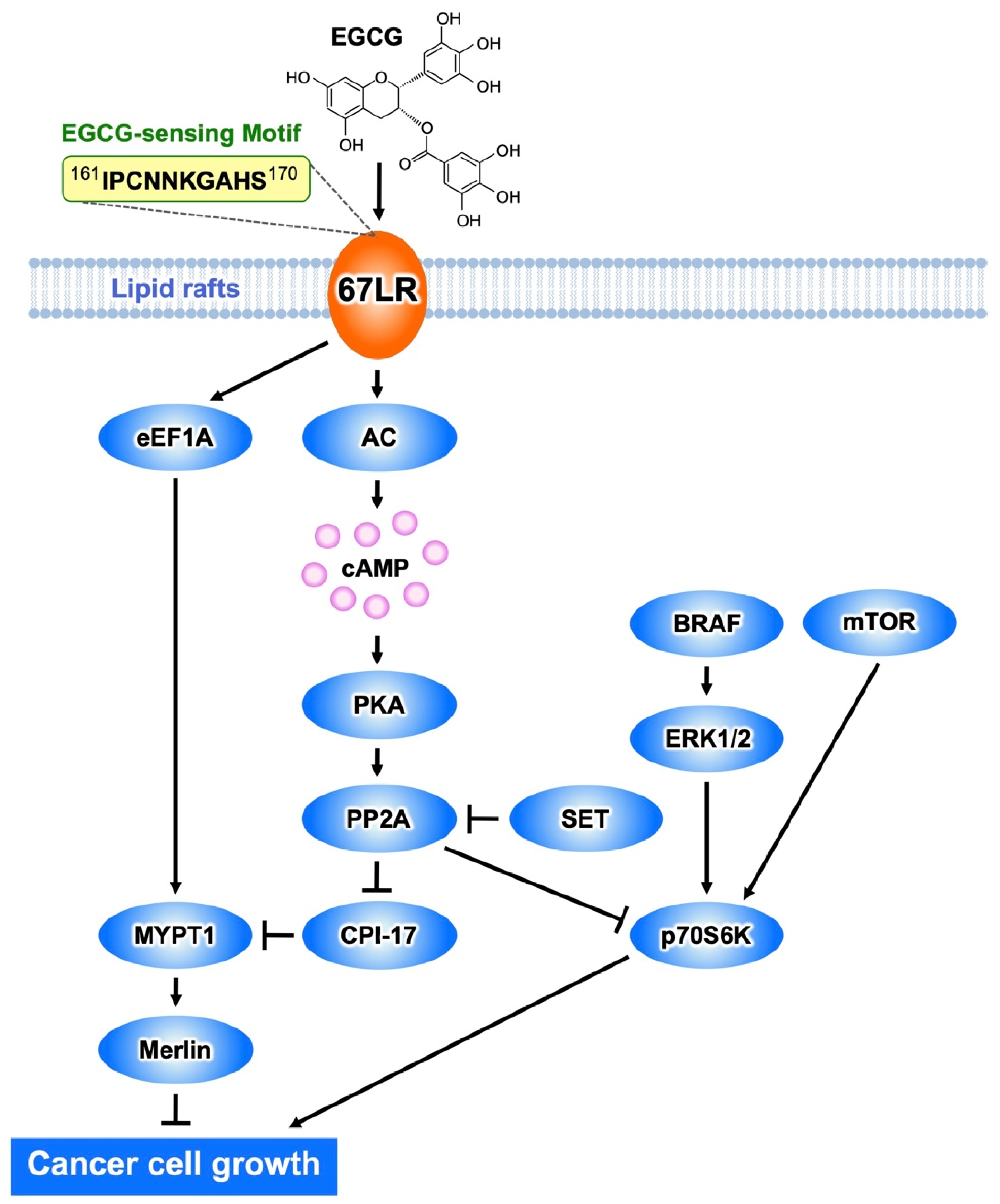
Figure 1. The EGCG-sensing pathway inhibits melanoma cell growth and proliferation by a mechanism mediated by 67LR. The interaction EGCG−67LR results in the activation of MYPT1 via AC/cAMP/PP2A- or the eEF1A-dependent pathway. Consequently, merlin is activated, and dephosphorylates MRLC. BRAF/ERK or mTOR-mediated activation of p70S6K is inhibited by PP2A.
In vivo studies confirmed that 67LR functions as a receptor of EGCG, which is critical to its anti-cancer effect. Thus, EGCG treatment substantially inhibited tumor growth in mice implanted with B16 cells harboring a control short hairpin RNA (shRNA), but not in those implanted with 67LR-ablated B16 cells [9].
Furthermore, 67LR is associated with adherence of tumor cells, migration, invasion, and metastasis [10]. It is also a target molecule for pathogenic prion proteins [11], Sindbis virus [12], and dengue virus [13]. Caffeine and other catechins (C, EC, EGC, and quercetin) do not bind to the surface of 67LR-expressing cells nor affect their growth at the concentration of 5 μM [5]. Galloylated, but not non-galloylated, catechins attach to the cancer cells. The binding ability of pyrogallol-type catechins (EGCG and GCG) is higher than that of catechol-type catechins (ECG and CG) [14], in correlation with their biological activities. Downregulation of the expression of 67LR reduces the activity of galloylated catechins. Thus, both the galloyl moiety and B-ring hydroxylation may be involved in biological activities of these compounds mediated by 67LR.
Strictinin, one of esters of gallic acid with a polyol, is a hydrolysable ellagitannin in green tea. It suppresses interleukin (IL)-4 signaling [15] by interacting with the non-lipid raft-associated IL-4 receptor (IL-4R) on the surface of the cell membrane [16]. Downregulation of 67LR does not interfere with this effect. The Kd value of IL-4R-strictinin complex is 4.53 μM, whereas that of complex of IL-4R with EGCG, which is mostly located in the lipid-raft region, is about 155 μM [16]. Thus, both the flavan-3-ol structure and galloyl moiety might play a crucial role in the interaction between 67LR and EGCG.
Recently, researchers revealed that EGCG formed oligomers by binding to 67LR [17]. Oligomer formation occurs after absorption, possibly in a manner similar to that of highly oligomerized procyanidin. OurThe data indicate a possible mechanism underlying the strong in vivo biological activities of EGCG.
Procyanidins are one of the well-known bioactive polyphenols in grape seeds and apple peels. Recent studies showed that these compounds have anti-cancer properties [18][19]. For instance, procyanidin C1 (PC1), an EC trimer, has anti-cancer effects [19]; however, its molecular mechanisms underlying this effect remain unknown. PC1 also binds to 67LR with a Kd value of 2.8 µM [20]. This interaction results in the activation of protein kinase A (PKA) and protein phosphatase 2A (PP2A). Consequently, it inhibits the growth of melanoma cells, by downregulation of phosphorylation levels of the C-kinase potentiated protein phosphatase-1 inhibitor protein of 17-kDa (CPI17) and dephosphorylation of myosin regulatory light chain (MRLC) proteins, followed by actin cytoskeleton remodeling. In the same study, catechin dimers procyanidin B1 (PB1) and procyanidin B2 (PB2) showed lack of cell surface binding with no significant activity on phosphorylation levels of MRLC in melanoma cells. Taken together, PC1 has anti-melanoma effects mediated by a 67LR-dependent mechanism. The mechanisms by which other EC oligomers interact with 67LR, such as tetramers and pentamers, remain unknown and further studies are needed.
2. Inhibitory Actions of EGCG on Cancer Cell Growth Mediated by 67LR
Melanoma is the deadliest skin cancer and is notorious for its resistance to treatment, but recent clinical trials of targeted therapies have shown promise [21][22]. On the other hand, many anti-cancer drugs are natural products or their derivatives, pointing to the usefulness of natural products in drug discovery. Functional genetic screening identified eukaryotic elongation factor 1A (eEF1A) as a critical anti-proliferative factor for melanoma cells [9]. eEF1A regulates various cellular processes, including the translation process in eukaryotes. EGCG substantially inhibited tumor growth in mice inoculated with mouse melanoma cells harboring a control shRNA, but not in mice inoculated with eEF1A-ablated melanoma cells, highlighting the role of eEF1A plays in the EGCG-induced anti-cancer effect.
It has been mentioned above that EGCG binding to 67LR results in cell growth inhibition as a result of a suppressing effect on the phosphorylation of MRLC at Thr18/Ser19 [23]. This is determined by EGCG-induced downregulation of the phosphorylation level of the myosin phosphatase target subunit 1 (MYPT1) at Thr696. Consequently, myosin phosphatase activity is upregulated.
EGCG administration substantially inhibited tumor volume increase in mice inoculated with B16 cells harboring a control shRNA, but not in mice inoculated with MYPT1-knockdowned B16 cells, indicating that MYPT1 is also an essential element for EGCG-induced cancer prevention. In eEF1A- or 67LR-knockdowned mouse melanoma cells, EGCG does not induce downregulation of the phosphorylation level of MYPT1Thr696 at the concentration of 1 μM. These findings indicate that MYPT1 plays a crucial role in EGCG signaling from both 67LR and eEF1A (Figure 1).
Abnormal activation of BRAF is the most frequently observed mutation event in patients with melanoma and is associated with constitutive hyperproliferation. Selective BRAF inhibition improves the outcomes of patients with mutated BRAF melanoma. Unfortunately, acquired resistance to BRAF inhibition is the problem. Therefore, there is an urgent need to identify new strategies of treatment of melanoma. EGCG (5 μM) inhibits melanoma cell growth independent of BRAF inhibitor sensitivity by regulating the activity of PP2A. Functional genetic screening identified this enzyme as a key player in lowering the growth of melanoma cells [24]. EGCG binding to 67LR elicits activation of PP2A via the adenylate cyclase (AC)/cyclic adenosine monophosphate (cAMP) axis, causing melanoma-specific mammalian target of rapamycin (mTOR) inhibition and merlin activation related to tumor suppression (Figure 1). Furthermore, the inhibition of mTOR causes strong synergism with the BRAF inhibitor in BRAF inhibitor-resistant melanoma. Additionally, Suvar3–9, an enhancer-of-zeste trithorax (SET), an oncoprotein that inhibits the activity of PP2A, is overexpressed in melanoma. EGCG (5 μM) activates PP2A and inhibits cell growth without affecting SET expression in melanoma. SET silencing significantly enhances 67LR/PP2A signaling in vivo and, consequently, the EGCG-elicited anti-melanoma activity [25]. Therefore, SET is expected to be an important target molecule for EGCG to potentiate its anti-melanoma action.
3. Cancer Cell Killing Effects of EGCG Mediated by 67LR
Green tea consumption has shown beneficial effects in patients with oral cancer [26], colorectal adenomas [27], prostate cancer [28], and early-stage chronic lymphocytic leukemia [29]. In a Phase II clinical trial in patients with chronic lymphocytic leukemia, Polyphenon ETM, containing 60% EGCG, the first botanical drug approved by the US Food and Drug Administration for the treatment of patients with external genital and perianal warts, showed a strong anti-cancer effect without causing serious side effects [29].
EGCG (5 or 10 μM) induces growth arrest and apoptosis in multiple myeloma (MM) [30] and acute myeloid leukemia (AML) cells [31] derived from cell lines and human patients; however, it has no impact on the growth of normal cells such as normal human peripheral blood mononuclear cells (PBMCs). The levels of 67LR expression are strongly upregulated in myeloma cells compared with PBMCs. The specific pro-apoptotic effect of EGCG on MM cells is prevented by RNAi-mediated inhibition of 67LR expression, further highlighting the importance of this receptor in the cancer killing effect of EGCG.
Changes in the clustering of lipid rafts activate various cellular signaling pathways, including those involved in apoptosis. EGCG (5 μM), but not biologically inactive EC (10 μM), induced clustering of lipid rafts in MM cells [32]. An anti-67LR antibody treatment blocked EGCG-elicited clustering of lipid rafts, whereas control antibody treatment did not. Lipid rafts are microdomains enriched in sphingolipids and cholesterol, and changes in their quantity cause changes in the integrity and function of lipid rafts. Cholesterol pretreatment impeded EGCG-induced lipid raft clustering and apoptosis in MM cells, indicating that lipid raft clustering is indispensable for the apoptotic cell death induced by EGCG.
Acid sphingomyelinase (ASM) has an essential role in lipid raft clustering through the production of ceramide. The expression of ASM is drastically upregulated in myeloid cells compared to its normal counterpart [24]. EGCG (5 μM) induced ASM activation in myeloid cells, but did not affect its normal counterpart [32]. Furthermore, an anti-67LR antibody treatment neutralized EGCG-elicited ASM activation, indicating that 67LR acts as a critical player in the effect of EGCG. Therefore, EGCG activates the sphingolipid cascade by eliciting the ASM/67LR axis, and this enzyme is indispensable for EGCG-elicited apoptosis by clustering of lipid rafts in MM cells (Figure 2).
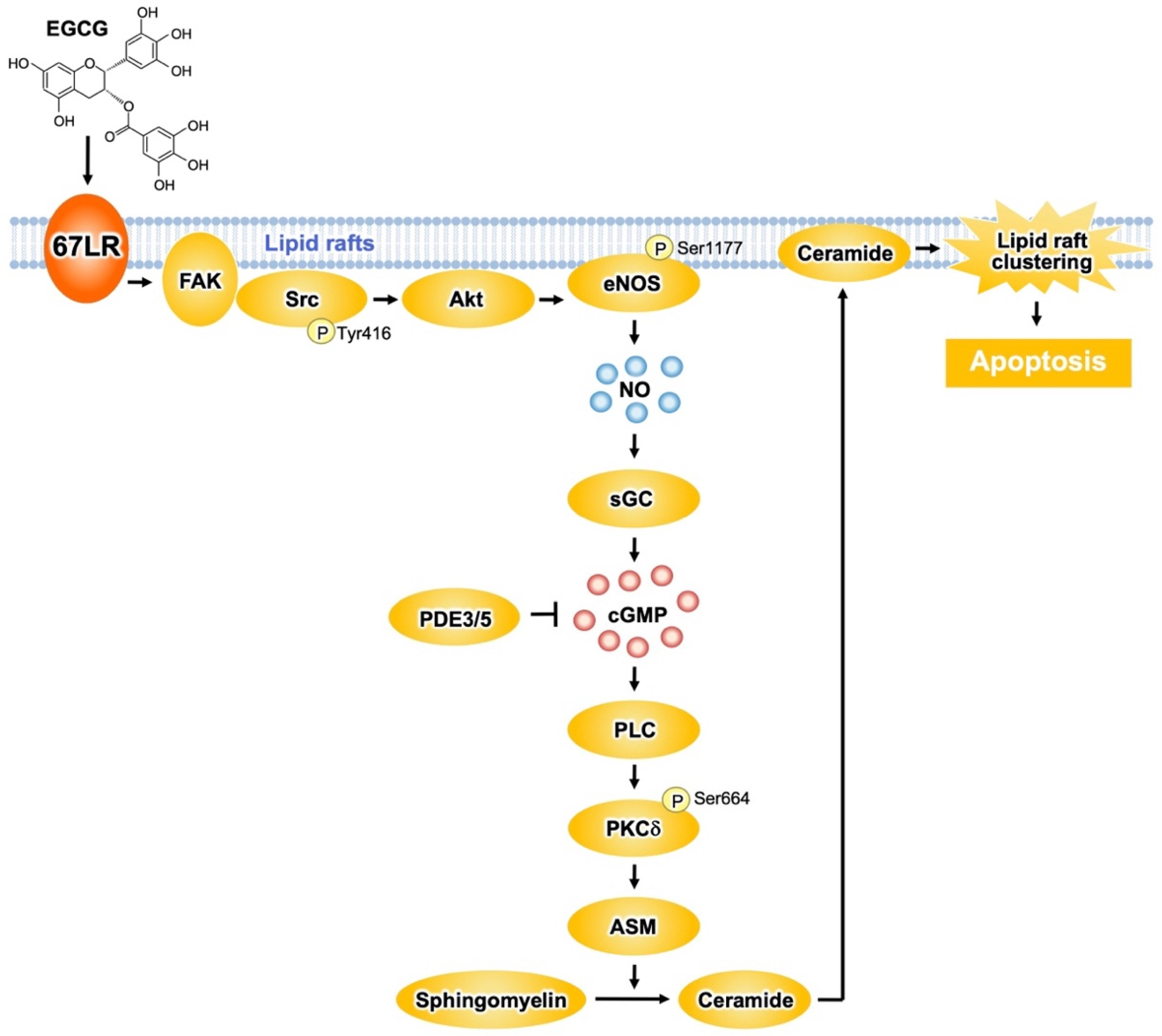
Figure 2. The 67LR-dependent EGCG-sensing pathway involved in multiple myeloma (MM) cell death. EGCG induces apoptosis in multiple myeloma (MM) cells via the activation of the 67LR/ FAK/Src/Akt/eNOS/NO/sGC/cGMP/ PLC/PKCδ/ASM signaling pathway.
Protein kinase Cδ (PKCδ) plays an essential role in the apoptosis-inducing pathway. EGCG (1 μM) upregulated the phosphorylation levels of PKCδSer664 in myeloid cells, without affecting its normal counterpart [32]. Pretreatment of the cells with an anti-67LR antibody inhibits the elevation of phosphorylation levels of PKCδSer664 induced by EGCG. Moreover, pharmacological inhibition of PKCδ neutralized the EGCG-induced ASM activation. Knockdown of ASM in myeloma cells had no influence on EGCG-elicited upregulation of phosphorylation levels of PKCδSer664. Thus, ASM activation elicited by EGCG may be a subordinate event that is triggered after upregulation of PKCδ activity. Oral intake of EGCG induced the activation of caspase 3, the determining step of apoptosis signaling, in myeloid cells. In addition, EGCG upregulated the phosphorylation levels of PKCδSer664 and increased ASM activity in cancer cells, suggesting that EGCG elicits the PKCδ/ASM axis in myeloid cells in vivo [32] (Figure 2).
67LR contributes to shear stress-elicited expression of nitric oxide synthase 3 (also endothelial nitric oxide synthase (eNOS)) [33]. EGCG (5 μM) increased NO levels in myeloid cells, but not in PBMCs from healthy donors [34]. This increase results from eNOS activation owing to phosphorylation at Ser1177 by Akt. Anti-67LR antibody pretreatment in myeloid cells inhibited eNOSSer1177 phosphorylation and prevented the increase in Akt kinase activity induced by EGCG (5 μM) [34]. Therefore, EGCG elicits the upregulation of NO levels by activating the 67LR/Akt/eNOS axis (Figure 2).
NO upregulates the levels of cyclic guanosine monophosphate (cGMP) via soluble guanylate cyclase (sGC)-dependent mechanisms. EGCG (5 μM) upregulates cGMP levels in myeloid cells without affecting its normal counterpart [34]. Anti-67LR antibody or sGC inhibitor pretreatment prevented EGCG-induced cGMP elevation. The pharmacological inhibition of sGC also attenuated EGCG-induced apoptosis and activation of ASM. Altogether, these data indicate that activation of the 67LR/cGMP signaling axis is the critical event for EGCG-induced apoptosis (Figure 2). Other catechins did not have any influence on cGMP levels.
EGCG elicits apoptotic cell death in several types of malignant cells though the induction of cGMP. However, the upstream mechanisms that occur after cGMP induction are not completely understood. It is demonstrated that a cGMP increase determined by EGCG induced PKCδSer664 phosphorylation and, consequently, ASM activation [35]. EGCG upregulated phospholipase C (PLC) activity in a similar manner to the treatment with cGMP inducers. PLC is involved in the production of diacylglycerol (DAG), which is required for PKCδ activation. Pharmacological inhibition of PLC prevents EGCG-induced ASM activation. Pharmacological inhibition of DAG strongly increases the activity of EGCG. Collectively, these findings indicate that EGCG induces apoptotic cell death in MM cells by activating the cGMP/PLC/PKCδ/ASM signaling pathway [35] (Figure 2).
The proto-oncogene c-Src greatly contributes to the anti-cancer effect of EGCG [36]. EGCG promotes c-SrcTyr416 phosphorylation, which is essential for the activity of the enzyme. Focal adhesion kinase (FAK), a regulator of Src phosphorylation, is colocalized with 67LR, and EGCG enhances the interaction between FAK and 67LR. Furthermore, pharmacological inhibition of Src and FAK prevents the early mechanisms in EGCG-elicited apoptosis: increase of Akt activity, upregulation of cGMP levels, and enhance of ASM activity. These observations suggest that FAK/Src highly contributes to the upstream signaling of EGCG-induced Akt/cGMP/ASM activation (Figure 2).
4. Anti-Inflammatory and Anti-Allergic Actions of EGCG Mediated by 67LR
Lipopolysaccharide (LPS) is one of the major components of Gram-negative bacteria, is a ligand of Toll-like receptor 4 (TLR4) signaling, and an inducer of inflammatory cytokines, which are involved in the onset and pathological development of septic shock. EGCG prevents LPS-elicited sepsis in mice and suppresses the inflammatory cytokines from macrophages [37]. Knockdown of 67LR impeded the anti-inflammatory effect of EGCG (1 μM) in mouse macrophages [38]. In addition, EGCG reduced TLR4 levels though 67LR-dependent mechanisms. It is known that overexpression of the Toll-interacting protein (Tollip) inhibits the activation of nuclear factor kappa B (NF-κB) and mitogen-activated protein kinases (MAPKs) in response to IL-1, TLR2, and TLR4 ligands [39]. Thus, Tollip negatively regulates the TLR-mediated signaling pathway. EGCG rapidly increased the level of Tollip and this effect is neutralized by 67LR knockdown. Therefore, 67LR is essential in manifesting the anti-inflammatory effect of EGCG and its ability to upregulate Tollip expression [40] (Figure 3). Peptidoglycan (PGN) is one of the pathogen-associated components derived from Gram-positive bacteria and strongly activates TLR2 signaling, whereas EGCG inhibits its effect by a mechanism involving 67LR and Tollip [40].
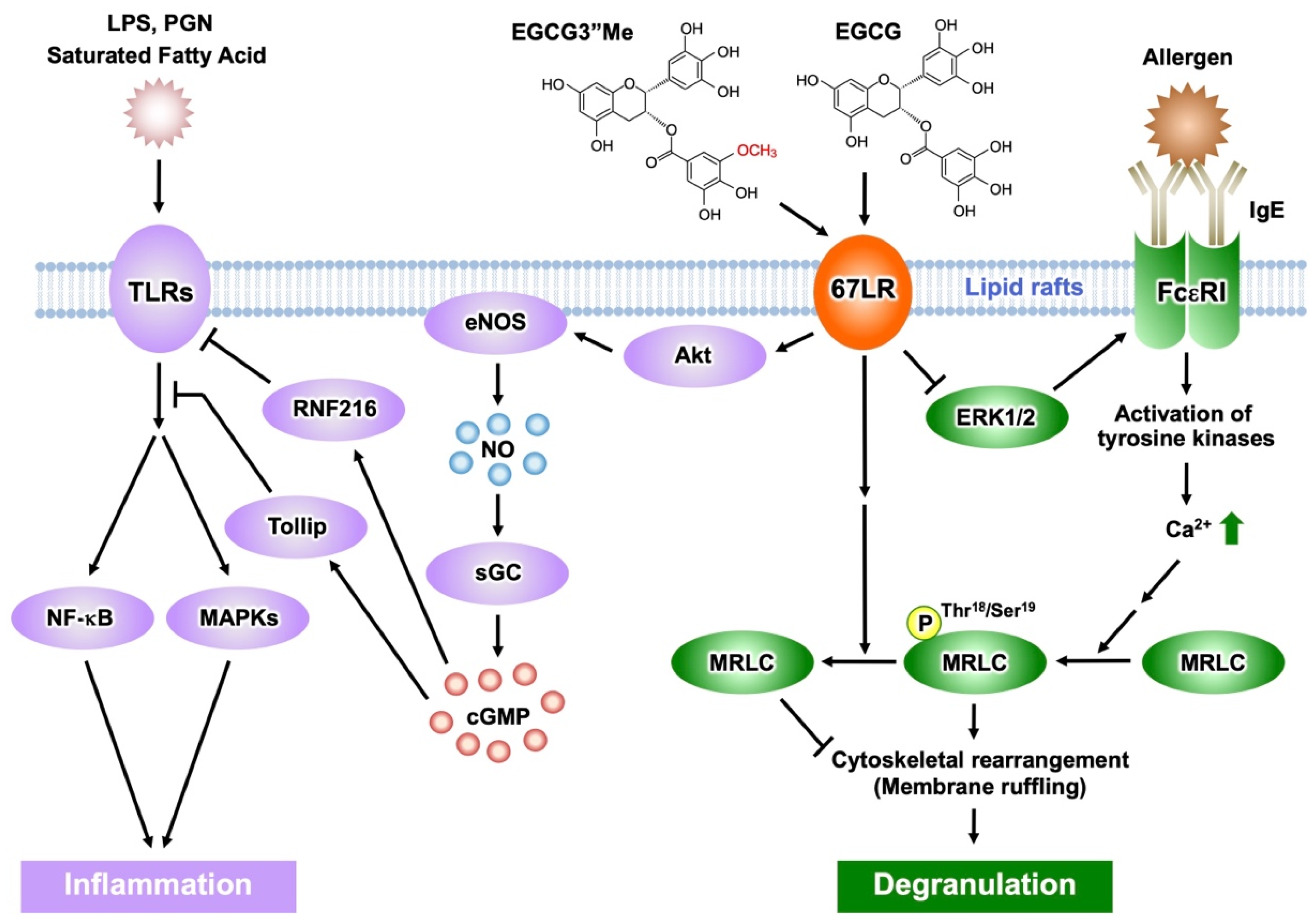
Figure 3. Activation of EGCG-sensing pathway following its binding to 67LR results in anti-inflammatory and anti-allergic actions. The anti-inflammatory effect of EGCG and EGCG3″Me results from inhibition of TLR signaling mediated by 67LR, Tollip, and RNF216. EGCG/EGCG3″Me binding to 67LR suppresses MRLC phosphorylation and the histamine release from basophils. EGCG/EGCG3″Me also inhibits ERK1/2 phosphorylation, reducing FcεRI expression.
Furthermore, EGCG (2.5 μM) increased Tollip expression by downregulation of E74-like ETS factor 1 (Elf-1) [41] via PP2A/cGMP-dependent mechanisms. These results were further confirmed by in vivo studies: oral administration of EGCG and induction of cGMP, elevated expression of Tollip, upregulation of cGMP levels in macrophages, and downregulated Elf-1 expression. cGMP acted as a key molecule in 67LR-dependent upregulation of the E3 ubiquitin-protein ring finger protein 216 (RNF216) (Figure 3). Additionally, researchers demonstrated that EGCG downregulated TLR4 levels by upregulating RNF216 [42].
EGCG and the cGMP inducer are attractive negative regulators capable of attenuating TLR4 signaling. Aberrant activation of TLR4 plays an essential role in inflammation caused by obesity, which is involved in several disorders (e.g., hypertriglyceridemia, hyperinsulinemia, and cardiovascular disease). Moreover, TLR4 expression in adipose tissues is reduced by a highly absorbable 67LR agonist, (−)-epigallocatechin-3-O-(3-O-methyl)-gallate (EGCG3″Me) at the concentration of 1 μM. It completely inhibits obesity-elicited overexpression of inflammatory cytokines, such as monocyte chemoattractant protein-1 and tumor necrosis factor-α. The intake of EGCG3″Me inhibited obesity-induced hyperinsulinemia and hypertriglyceridemia. Collectively, 67LR is a potent target for relieving inflammation caused by obesity.
Mast cells and basophils mediate immunoglobulin (Ig) E-induced immediate allergic reactions. Allergen binding to IgE bound to the high-affinity IgE receptor (FcεRI) on the surface of mast cells or basophils leads to the release of preformed and newly generated inflammatory mediators, including histamine. Such mediators are responsible for the various symptoms of allergic reactions, such as atopic dermatitis, bronchial asthma, and food allergy [43][44]. The early phase of mast cell and basophil activation includes activation of protein tyrosine kinases and their substrates, an increase in the concentration of secondary messengers such as inositol trisphosphate and DAG, and elevation of intracellular Ca2+ levels [45][46]. The late phase of activation is caused after the influx of Ca2+ and includes the fusion of secretory granules with the cell membrane, remodeling of the actin cytoskeleton, and radical morphological changes [47][48][49]. EGCG inhibited calcium ionophore A23187-induced histamine release from human basophilic KU812 cells, but could not inhibit the elevation of intracellular Ca2+ levels after stimulation with A23187 [50]. Therefore, the effect of EGCG on histamine release will occur after the intracellular Ca2+ concentration has increased. Phosphorylation of MRLCThr18/Ser19 temporally correlates with degranulation in rat basophilic RBL-2H3 cells, while its inhibition impairs degranulation [51]. EGCG, but not EGC, inhibits histamine release and reduces the concentration of phosphorylated MRLC [50]. The effect of EGCG (25 μM) on histamine release or the phosphorylation of MRLCThr18/Ser19 was inhibited by treating with anti-67LR antibody or downregulating 67LR expression [50]. These results suggest that the inhibitory effect of EGCG on degranulation is induced by modifying the myosin cytoskeleton through the binding of EGCG to the cell surface 67LR. When basophilic cells were stimulated with A23187 in the presence of EGCG (25 μM), membrane ruffling, one effect of actin remodeling, was inhibited and biased F-actin accumulation was caused. Moreover, EGCG-induced actin remodeling was abrogated in both anti-67LR antibody-treated cells and 67LR-ablated cells [50]. These results indicate that EGCG-induced actin remodeling is caused by a reduction in MRLC Thr18/Ser19 phosphorylation, mediated by the binding of EGCG to 67LR. Therefore, cytoskeletal remodeling may play an essential role in the inhibitory effect of EGCG on histamine release.
FcεRI is involved in the induction and maintenance of IgE-mediated allergic responses. In FcεRIα chain-deficient mice, IgE cannot bind to the surface of mast cells, thereby inhibiting the induction of degranulation [52]. Thus, downregulation of FcεRI expression in mast cells and basophils may contribute to attenuation of IgE-mediated allergic symptoms. It has been reported that EGCG (25 μM) can decrease the cell surface expression of FcεRI in KU812 cells, by inhibiting the phosphorylation of extracellular signal-regulated kinase1/2 (ERK1/2) [8]. Furthermore, the disruption of lipid rafts prevented the inhibitory effect of EGCG on ERK1/2. Thus, the interaction between EGCG and lipid rafts is important for the ability of EGCG to suppress FcεRI expression, and ERK1/2 may contribute to this suppression signal. Researchers also revealed that the knockdown of 67LR inhibited the effect of EGCG on FcεRI expression. Moreover, the inhibitory effect of EGCG on the phosphorylation of ERK1/2 was abrogated in 67LR-ablated cells. OurThe findings indicate that the effect of EGCG on ERK1/2 phosphorylation correlates with 67LR expression, which implies that 67LR is the molecule responsible for the transduction of EGCG’s downregulatory signaling on FcεRI (Figure 3).
The O-methylated derivatives of EGCG, EGCG3″Me, and (−)-epigallpcatechin-3-O-(4-O-methyl)-gallate (EGCG4″Me), which were isolated from tea leaves such as Tong-ting oolong or Benifuuki tea cultivars, inhibit allergic reactions in vitro [53][54]. The in vivo anti-allergic effects of O-methylated EGCG, assessed using mouse models of type I and IV allergic reactions, were stronger than those of EGCG [53][55]. Mast cell activation was strongly inhibited by these methylated catechins, mediated through preventing tyrosine phosphorylation of cellular proteins, histamine/leukotriene release, and IL-2 secretion after FcεRI cross-linking [56]. A double-blind clinical trial assessed the efficacy of EGCG3″Me-rich Benifuuki green tea on allergic cedar pollinosis. Drinking 1.5 g of tea powder with water twice a day for 13 weeks improved the symptomatology of patients [57]. EGCG3″Me (25 μM) inhibited the release of histamine and suppressed the expression of FcεRI in KU812 cells, similar to EGCG [54][58]. The activity of EGCG3″Me was decreased by RNAi-mediated knockdown of 67LR expression [59]. Therefore, the anti-histaminic effect of EGCG3″Me is mediated by suppression of MRLC phosphorylation following 67LR binding. 67LR also mediates the suppressive effect of EGCG3″Me on FcεRI expression by reducing ERK1/2 phosphorylation.
EGCG is not stable and easily degraded in vivo. On the other hand, EGCG3″Me and EGCG4″Me are absorbed efficiently and are more stable than EGCG in animal and human plasma. This could explain the potent in vivo anti-allergic activities of O-methylated EGCG. EGCG undergoes methylation, and (−)-4′-O-methyl-epigallocatechin-3-O-(4-O-methyl) gallate (EGCG4′4″diMe) is a major metabolite of EGCG detected in plasma [60]. It did not demonstrate a suppressive effect on KU812 cells at the concentration of 25 μM [61]. OurThe findings of O-methylated EGCGs may lead to the understanding of the physiological activities of EGCG in vivo, but further study on the relationship between EGCG metabolites and 67LR is required.
5. MicroRNA-Mediated Anti-Cancer Effect of EGCG via 67LR
MicroRNAs (miRNAs) are small single-stranded, non-coding RNAs that regulate gene expression by inducing mRNA degradation or translational inhibition [62]. They play critical roles in various biological events, including cell proliferation [63], apoptosis [64], differentiation [65], inflammation [66], and metabolism [67], and are implicated in various diseases [68][69]. Some miRNAs are aberrantly expressed in various types of cancer cells and their dysregulation leads to cancer progression [70][71][72]. Therefore, modulating miRNA activity by dietary polyphenols may be an effective strategy against cancer [73][74][75][76]. Among the diverse polyphenols, some studies have investigated whether flavonoids similar to EGCG, belonging to flavan-3-ols, have such miRNA-mediated effects.
Isoflavones are a group of flavonoids that are found predominantly in soy and soy products. Their effects are mediated by estrogen receptors (ER). Equol is an intestinal metabolite of the representative soy isoflavone daidzein. At the concentration of 10 μM, it inhibits the proliferation of human cervical cancer HeLa cells and mouse melanoma B16 cells in an ER-independent manner. Its target may be the non-canonical poly(A) polymerase, PAP-associated domain containing 5 (PAPD5) [77]. Thus, oral administration of equol suppressed tumor growth in control mice inoculated with B16 cells, but not in mice inoculated with PAPD5-ablated B16 cells. Furthermore, it promoted tumor growth in PAPD5-ablated human breast cancer MCF-7 cells with high expression of ERα and induced polyadenylation of snoRNAs in a PAPD5-dependent manner. The expression of miR-320a in tumors was upregulated by its oral administration. Consequently, equol may show a dual effect on ER-positive cancer cells: it inhibits proliferation, an effect mediated by PAPD5 via miR-320a and enhances proliferation, an effect mediated by ERα.
The effects of delphinidin, a flavonoid belonging to anthocyanidins, on the levels of muscle-specific RING-finger protein 1 (MuRF1), miR-23a, and NFATc3 were assessed by in vitro and in vivo studies [78]. Delphinidin (5 μM) inhibits dexamethasone-induced upregulation of MuRF1 expression and downregulation of miR-23a and NFATc3 in mouse myoblast C2C12 cells. Its oral administration prevented the reduction of muscle mass in the gastrocnemius muscle, while reducing MuRF1 and increasing miR-23a and NFATc3 expression. This mechanism of action indicates that delphinidin intake may exert a preventive effect on disuse muscle atrophy.
Modulating miRNAs by flavonoids might be involved in their pharmacological properties. However, the effects of EGCG on miRNA expression in melanoma cells remain unclear. To address this, researchers conducted an miRNA microarray analysis [79]. EGCG (10 μM) upregulated the expression of miRNA-let-7b in melanoma cells by activation of the 67LR-dependent cAMP/PKA/PP2A signaling pathway. EGCG-induced upregulation of let-7b downregulated high-mobility group A2 (HMGA2), a target gene related to tumor progression. These observations offer a new insight on the mechanisms for EGCG-induced regulation of miRNA. The fact that EGCG can regulate miRNA via 67LR will provide a deeper understanding of the 67LR-mediated EGCG-sensing systems.
References
- Chen, Z.Y.; Zhu, Q.Y.; Tsang, D.; Huang, Y. Degradation of Green Tea Catechins in Tea Drinks. J. Agric. Food Chem. 2001, 49, 477–482.
- Howells, L.M.; Moiseeva, E.P.; Neal, C.P.; Foreman, B.E.; Andreadi, C.K.; Sun, Y.Y.; Hudson, E.A.; Manson, M.M. Predicting the Physiological Relevance of in Vitro Cancer Preventive Activities of Phytochemicals. Acta Pharmacol. Sin. 2007, 28, 1274–1304.
- Yang, C.S.; Wang, X.; Lu, G.; Picinich, S.C. Cancer Prevention by Tea: Animal Studies, Molecular Mechanisms and Human Relevance. Nat. Rev. Cancer 2009, 9, 429–439.
- Negri, A.; Naponelli, V.; Rizzi, F.; Bettuzzi, S. Molecular Targets of Epigallocatechin—Gallate (EGCG): A Special Focus on Signal Transduction and Cancer. Nutrients 2018, 10, 1936.
- Tachibana, H.; Koga, K.; Fujimura, Y.; Yamada, K. A Receptor for Green Tea Polyphenol EGCG. Nat. Struct. Mol. Biol. 2004, 11, 380–381.
- Fujimura, Y.; Sumida, M.; Sugihara, K.; Tsukamoto, S.; Yamada, K.; Tachibana, H. Green Tea Polyphenol EGCG Sensing Motif on the 67-KDa Laminin Receptor. PLoS ONE 2012, 7, e37942.
- Fujimura, Y.; Yamada, K.; Tachibana, H. A Lipid Raft-Associated 67 KDa Laminin Receptor Mediates Suppressive Effect of Epigallocatechin-3-O-Gallate on FcεRI Expression. Biochem. Biophys. Res. Commun. 2005, 336, 674–681.
- Fujimura, Y.; Tachibana, H.; Yamada, K. Lipid Raft-Associated Catechin Suppresses the FcεRI Expression by Inhibiting Phosphorylation of the Extracellular Signal-Regulated Kinase1/2. FEBS Lett. 2004, 556, 204–210.
- Umeda, D.; Yano, S.; Yamada, K.; Tachibana, H. Green Tea Polyphenol Epigallocatechin-3-Gallate Signaling Pathway through 67-KDa Laminin Receptor. J. Biol. Chem. 2008, 283, 3050–3058.
- Mafune, K.; Ravikumar, T.S. Anti-Sense RNA of 32-KDa Laminin-Binding Protein Inhibits Attachment and Invasion of a Human Colon Carcinoma Cell Line. J. Surg. Res. 1992, 52, 340–346.
- Gauczynski, S.; Peyrin, J.M.; Haïk, S.; Leucht, C.; Hundt, C.; Rieger, R.; Krasemann, S.; Deslys, J.P.; Dormont, D.; Lasmézas, C.I.; et al. The 37-KDa/67-KDa Laminin Receptor Acts as the Cell-Surface Receptor for the Cellular Prion Protein. EMBO J. 2001, 20, 5863–5875.
- Wang, K.S.; Kuhn, R.J.; Strauss, E.G.; Ou, S.; Strauss, J.H. High-Affinity Laminin Receptor Is a Receptor for Sindbis Virus in Mammalian Cells. J. Virol. 1992, 66, 4992–5001.
- Thepparit, C.; Smith, D.R. Serotype-Specific Entry of Dengue Virus into Liver Cells: Identification of the 37-Kilodalton/67-Kilodalton High-Affinity Laminin Receptor as a Dengue Virus Serotype 1 Receptor. J. Virol. 2004, 78, 12647–12656.
- Fujimura, Y.; Umeda, D.; Yamada, K.; Tachibana, H. The Impact of the 67 KDa Laminin Receptor on Both Cell-Surface Binding and Anti-Allergic Action of Tea Catechins. Arch. Biochem. Biophys. 2008, 476, 133–138.
- Tachibana, H.; Kubo, T.; Miyase, T.; Tanino, S.; Yoshimoto, M.; Sano, M.; Yamamoto-Maeda, M.; Yamada, K. Identification of an Inhibitor for Interleukin 4-Induced ε Germline Transcription and Antigen-Specific IgE Production In Vivo. Biochem. Biophys. Res. Commun. 2001, 280, 53–60.
- Kim, Y.H.; Ninomiya, Y.; Yamashita, S.; Kumazoe, M.; Huang, Y.; Nakahara, K.; Won, Y.S.; Murata, M.; Fujimura, Y.; Yamada, K.; et al. IL-4 Receptor α in Non-Lipid Rafts Is the Target Molecule of Strictinin in Inhibiting STAT6 Activation. Biochem. Biophys. Res. Commun. 2014, 450, 824–830.
- Huang, Y.; Sumida, M.; Kumazoe, M.; Sugihara, K.; Suemasu, Y.; Yamada, S.; Yamashita, S.; Miyakawa, J.; Takahashi, T.; Tanaka, H.; et al. Oligomer Formation of a Tea Polyphenol, EGCG, on Its Sensing Molecule 67 KDa Laminin Receptor. Chem. Commun. 2017, 53, 1941–1944.
- Gossé, F.; Guyot, S.; Roussi, S.; Lobstein, A.; Fischer, B.; Seiler, N.; Raul, F. Chemopreventive Properties of Apple Procyanidins on Human Colon Cancer-Derived Metastatic SW620 Cells and in a Rat Model of Colon Carcinogenesis. Carcinogenesis 2005, 26, 1291–1295.
- Miura, T.; Chiba, M.; Kasai, K.; Nozaka, H.; Nakamura, T.; Shoji, T.; Kanda, T.; Ohtake, Y.; Sato, T. Apple Procyanidins Induce Tumor Cell Apoptosis through Mitochondrial Pathway Activation of Caspase-3. Carcinogenesis 2008, 29, 585–593.
- Bae, J.; Kumazoe, M.; Murata, K.; Fujimura, Y.; Tachibana, H. procyanidin c1 inhibits melanoma cell growth by activating 67-kda laminin receptor signaling. Mol. Nutr. Food Res. 2020, 64, 1900986.
- Salama, A.K.S.; Flaherty, K.T. Braf in melanoma: Current strategies and Future Directions. Clin. Cancer Res. 2013, 19, 4326–4334.
- Chin, L.; Garraway, L.A.; Fisher, D.E. Malignant Melanoma: Genetics and Therapeutics in the Genomic Era. Genes Dev. 2006, 20, 2149–2182.
- Umeda, D.; Tachibana, H.; Yamada, K. Epigallocatechin-3-O-Gallate Disrupts Stress Fibers and the Contractile Ring by Reducing Myosin Regulatory Light Chain Phosphorylation Mediated through the Target Molecule 67 KDa Laminin Receptor. Biochem. Biophys. Res. Commun. 2005, 333, 628–635.
- Tsukamoto, S.; Huang, Y.; Umeda, D.; Yamada, S.; Yamashita, S.; Kumazoe, M.; Kim, Y.; Murata, M.; Yamada, K.; Tachibana, H. 67-KDa Laminin Receptor-Dependent Protein Phosphatase 2A (PP2A) Activation Elicits Melanoma-Specific Antitumor Activity Overcoming Drug Resistance. J. Biol. Chem. 2014, 289, 32671–32681.
- Tsukamoto, S.; Huang, Y.; Kumazoe, M.; Lesnick, C.; Yamada, S.; Ueda, N.; Suzuki, T.; Yamashita, S.; Kim, Y.H.; Fujimura, Y.; et al. Sphingosine Kinase-1 Protects Multiple Myeloma from Apoptosis Driven by Cancer-Specific Inhibition of RTKs. Mol. Cancer Ther. 2015, 14, 2303–2312.
- Li, N.; Sun, Z.; Han, C.; Chen, J. The Chemopreventive Effects of Tea on Human Oral Precancerous Mucosa Lesions. Proc. Soc. Exp. Biol. Med. 1999, 220, 218–224.
- Shimizu, M.; Fukutomi, Y.; Ninomiya, M.; Nagura, K.; Kato, T.; Araki, H.; Suganuma, M.; Fujiki, H.; Moriwaki, H. Green Tea Extracts for the Prevention of Metachronous Colorectal Adenomas: A Pilot Study. Cancer Epidemiol. Biomark. Prev. 2008, 17, 3020–3025.
- Bettuzzi, S.; Brausi, M.; Rizzi, F.; Castagnetti, G.; Peracchia, G.; Corti, A. Chemoprevention of Human Prostate Cancer by Oral Administration of Green Tea Catechins in Volunteers with High-Grade Prostate Intraepithelial Neoplasia: A Preliminary Report from a One-Year Proof-of-Principle Study. Cancer Res. 2006, 66, 1234–1240.
- Shanafelt, T.D.; Call, T.G.; Zent, C.S.; Leis, J.F.; Laplant, B.; Bowen, D.A.; Roos, M.; Laumann, K.; Ghosh, A.K.; Lesnick, C.; et al. Phase 2 Trial of Daily, Oral Polyphenon e in Patients with Asymptomatic, Rai Stage 0 to II Chronic Lymphocytic Leukemia. Cancer 2013, 119, 363–370.
- Shammas, M.A.; Neri, P.; Koley, H.; Batchu, R.B.; Bertheau, R.C.; Munshi, V.; Prabhala, R.; Fulciniti, M.; Yu, T.T.; Treon, S.P.; et al. Specific Killing of Multiple Myeloma Cells by (−)-Epigallocatechin-3- Gallate Extracted from Green Tea: Biologic Activity and Therapeutic Implications. Blood 2006, 108, 2804–2810.
- Kumazoe, M.; Kim, Y.; Bae, J.; Takai, M.; Murata, M.; Suemasu, Y.; Sugihara, K.; Yamashita, S.; Tsukamoto, S.; Huang, Y.; et al. Phosphodiesterase 5 Inhibitor Acts as a Potent Agent Sensitizing Acute Myeloid Leukemia Cells to 67-KDa Laminin Receptor-Dependent Apoptosis. FEBS Lett. 2013, 587, 3052–3057.
- Tsukamoto, S.; Hirotsu, K.; Kumazoe, M.; Goto, Y.; Sugihara, K.; Suda, T.; Tsurudome, Y.; Suzuki, T.; Yamashita, S.; Kim, Y.; et al. Green Tea Polyphenol EGCG Induces Lipid-Raft Clustering and Apoptotic Cell Death by Activating Protein Kinase Cδ and Acid Sphingomyelinase through a 67 KDa Laminin Receptor in Multiple Myeloma Cells. Biochem. J. 2012, 443, 525–534.
- Gloe, T.; Riedmayr, S.; Sohn, H.Y.; Pohl, U. The 67-KDa Laminin-Binding Protein Is Involved in Shear Stress-Dependent Endothelial Nitric-Oxide Synthase Expression. J. Biol. Chem. 1999, 274, 15996–16002.
- Kumazoe, M.; Sugihara, K.; Tsukamoto, S.; Huang, Y.; Tsurudome, Y.; Suzuki, T.; Suemasu, Y.; Ueda, N.; Yamashita, S.; Kim, Y.; et al. 67-KDa Laminin Receptor Increases CGMP to Induce Cancer-Selective Apoptosis. J. Clin. Investig. 2013, 123, 787–799.
- Bae, J.; Kumazoe, M.; Takeuchi, C.; Hidaka, S.; Fujimura, Y.; Tachibana, H. Epigallocatechin-3-O-Gallate Induces Acid Sphingomyelinase Activation through Activation of Phospholipase C. Biochem. Biophys. Res. Commun. 2019, 520, 186–191.
- Kumazoe, M.; Kadomatsu, M.; Bae, J.; Otsuka, Y.; Fujimura, Y.; Tachibana, H. Src Mediates Epigallocatechin-3-O-Gallate-Elicited Acid Sphingomyelinase Activation. Molecules 2020, 25, 5481.
- Li, W.; Ashok, M.; Li, J.; Yang, H.; Sama, A.E.; Wang, H. A Major Ingredient of Green Tea Rescues Mice from Lethal Sepsis Partly by Inhibiting HMGB1. PLoS ONE 2007, 2, e1153.
- Hong Byun, E.; Fujimura, Y.; Yamada, K.; Tachibana, H. TLR4 Signaling Inhibitory Pathway Induced by Green Tea Polyphenol Epigallocatechin-3-Gallate through 67-KDa Laminin Receptor. J. Immunol. 2010, 185, 33–45.
- Takeda, K.; Akira, S. TLR Signaling Pathways. Semin. Immunol. 2004, 16, 3–9.
- Byun, E.H.; Omura, T.; Yamada, K.; Tachibana, H. Green Tea Polyphenol Epigallocatechin-3-Gallate Inhibits TLR2 Signaling Induced by Peptidoglycan through the Polyphenol Sensing Molecule 67-KDa Laminin Receptor. FEBS Lett. 2011, 585, 814–820.
- Kumazoe, M.; Yamashita, M.; Nakamura, Y.; Takamatsu, K.; Bae, J.; Yamashita, S.; Yamada, S.; Onda, H.; Nojiri, T.; Kangawa, K.; et al. Green Tea Polyphenol EGCG Upregulates Tollip Expression by Suppressing Elf-1 Expression. J. Immunol. 2017, 199, 3261–3269.
- Kumazoe, M.; Nakamura, Y.; Yamashita, M.; Suzuki, T.; Takamatsu, K.; Huang, Y.; Bae, J.; Yamashita, S.; Murata, M.; Yamada, S.; et al. Green Tea Polyphenol Epigallocatechin-3-Gallate Suppresses Toll-like Receptor 4 Expression via up-Regulation of E3 Ubiquitin-Protein Ligase RNF216. J. Biol. Chem. 2017, 292, 4077–4088.
- Ravetch, J.V.; Kinet, J.P. Fc Receptors. Annu. Rev. Immunol. 1991, 9, 457–492.
- Metzger, H. The Receptor with High Affinity for IgE. Immunol. Rev. 1992, 125, 37–48.
- Turner, H.; Kinet, J.P. Signalling through the High-Affinity IgE Receptor FcεRI. Nature 1999, 402, 24–30.
- Rivera, J. Molecular Adapters in FcεRI Signaling and the Allergic Response. Curr. Opin. Immunol. 2002, 14, 688–693.
- Pfeiffer, J.R.; Seagrave, J.C.; Davis, B.H.; Deanin, G.G.; Oliver, J.M. Membrane and Cytoskeletal Changes Associated with IgE-Mediated Serotonin Release from Rat Basophilic Leukemia Cells. J. Cell Biol. 1985, 101, 2145–2155.
- Choi, O.H.; Adelstein, R.S.; Beaven, M.A. Secretion from Rat Basophilic RBL-2H3 Cells Is Associated with Diphosphorylation of Myosin Light Chains by Myosin Light Chain Kinase as Well as Phosphorylation by Protein Kinase C. J. Biol. Chem. 1994, 269, 536–541.
- Edgar, A.J.; Bennett, J.P. Circular Ruffle Formation in Rat Basophilic Leukemia Cells in Response to Antigen Stimulation. Eur. J. Cell Biol. 1997, 73, 132–140.
- Fujimura, Y.; Umeda, D.; Kiyohara, Y.; Sunada, Y.; Yamada, K.; Tachibana, H. The Involvement of the 67 KDa Laminin Receptor-Mediated Modulation of Cytoskeleton in the Degranulation Inhibition Induced by Epigallocatechin-3-O-Gallate. Biochem. Biophys. Res. Commun. 2006, 348, 524–531.
- Ludowyke, R.I.; Peleg, I.; Beaven, M.A.; Adelstein, R.S. Antigen-Induced Secretion of Histamine and the Phosphorylation of Myosin by Protein Kinase C in Rat Basophilic Leukemia Cells. J. Biol. Chem. 1989, 264, 12492–12501.
- Dombrowicz, D.; Flamand, V.; Brigman, K.K.; Koller, B.H.; Kinet, J.P. Abolition of Anaphylaxis by Targeted Disruption of the High Affinity Immunoglobulin E Receptor α Chain Gene. Cell 1993, 75, 969–976.
- Sano, M.; Suzuki, M.; Miyase, T.; Yoshino, K.; Maeda-Yamamoto, M. Novel Antiallergic Catechin Derivatives Isolated from Oolong Tea. J. Agric. Food Chem. 1999, 47, 1906–1910.
- Tachibana, H.; Sunada, Y.; Miyase, T.; Sano, M.; Maeda-Yamamoto, M.; Yamada, K. Identification of a Methylated Tea Catechin as an Inhibitor of Degranulation in Human Basophilic KU812 Cells. Biosci. Biotechnol. Biochem. 2000, 64, 452–454.
- Suzuki, M.; Yoshino, K.; Maeda-Yamamoto, M.; Miyase, T.; Sano, M. Inhibitory Effects of Tea Catechins and O-Methylated Derivatives of (−)-Epigallocatechin-3-O-Gallate on Mouse Type IV Allergy. J. Agric. Food Chem. 2000, 48, 5649–5653.
- Maeda-Yamamoto, M.; Inagaki, N.; Kitaura, J.; Chikumoto, T.; Kawahara, H.; Kawakami, Y.; Sano, M.; Miyase, T.; Tachibana, H.; Nagai, H.; et al. O-Methylated Catechins from Tea Leaves Inhibit Multiple Protein Kinases in Mast Cells. J. Immunol. 2004, 172, 4486–4492.
- Maeda-Yamamoto, M.; Ema, K.; Shibuichi, I. In Vitro and in Vivo Anti-Allergic Effects of “benifuuki” Green Tea Containing O-Methylated Catechin and Ginger Extract Enhancement. Cytotechnology 2007, 55, 135–142.
- Fujimura, Y.; Tachibana, H.; Maeda-Yamamoto, M.; Miyase, T.; Sano, M.; Yamada, K. Antiallergic Tea Catechin, (−)-Epigallocatechin-3-O-(3-O-Methyl)-Gallate, Suppresses FcεRl Expression in Human Basophilic KU812 Cells. J. Agric. Food Chem. 2002, 50, 5729–5734.
- Fujimura, Y.; Umeda, D.; Yano, S.; Maeda-Yamamoto, M.; Yamada, K.; Tachibana, H. The 67 KDa Laminin Receptor as a Primary Determinant of Anti-Allergic Effects of O-Methylated EGCG. Biochem. Biophys. Res. Commun. 2007, 364, 79–85.
- Lambert, J.D.; Yang, C.S. Cancer Chemopreventive Activity and Bioavailability of Tea and Tea Polyphenols. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2003, 523–524, 201–208.
- Yano, S.; Fujimura, Y.; Umeda, D.; Miyase, T.; Yamada, K.; Tachibana, H. Relationship between the Biological Activities of Methylated Derivatives of (−)-Epigallocatechin-3-O-Gallate (EGCG) and Their Cell Surface Binding Activities. J. Agric. Food Chem. 2007, 55, 7144–7148.
- Ambros, V. MicroRNAs: Tiny Regulators with Great Potential. Cell 2001, 107, 823–826.
- Rathod, S.S.; Rani, S.B.; Khan, M.; Muzumdar, D.; Shiras, A. Tumor Suppressive MiRNA-34a Suppresses Cell Proliferation and Tumor Growth of Glioma Stem Cells by Targeting Akt and Wnt Signaling Pathways. FEBS Open Bio 2014, 4, 485–495.
- Zhao, L.; Gu, H.; Chang, J.; Wu, J.; Wang, D.; Chen, S.; Yang, X.; Qian, B. MicroRNA-383 Regulates the Apoptosis of Tumor Cells through Targeting Gadd45g. PLoS ONE 2014, 9, e110472.
- Rusca, N.; Dehò, L.; Montagner, S.; Zielinski, C.E.; Sica, A.; Sallusto, F.; Monticelli, S. MiR-146a and NF-ΚB1 Regulate Mast Cell Survival and T Lymphocyte Differentiation. Mol. Cell. Biol. 2012, 32, 4432–4444.
- Sun, X.; Icli, B.; Wara, A.K.; Belkin, N.; He, S.; Kobzik, L.; Hunninghake, G.M.; Vera, M.P.; Blackwell, T.S.; Baron, R.M.; et al. MicroRNA-181b Regulates NF-ΚB-Mediated Vascular Inflammation. J. Clin. Investig. 2012, 122, 1973–1990.
- Kornfeld, J.W.; Baitzel, C.; Könner, A.C.; Nicholls, H.T.; Vogt, M.C.; Herrmanns, K.; Scheja, L.; Haumaitre, C.; Wolf, A.M.; Knippschild, U.; et al. Obesity-Induced Overexpression of MiR-802 Impairs Glucose Metabolism through Silencing of Hnf1b. Nature 2013, 494, 111–115.
- Zou, Q.; Li, J.; Hong, Q.; Lin, Z.; Wu, Y.; Shi, H.; Ju, Y. Prediction of MicroRNA-Disease Associations Based on Social Network Analysis Methods. Biom. Res. Int. 2015, 2015, 810514.
- Zou, Q.; Li, J.; Song, L.; Zeng, X.; Wang, G. Similarity Computation Strategies in the MicroRNA-Disease Network: A Survey. Brief. Funct. Genom. 2016, 15, 55–64.
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA Expression Profiles Classify Human Cancers. Nature 2005, 435, 834–838.
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A MicroRNA Expression Signature of Human Solid Tumors Defines Cancer Gene Targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261.
- Parikh, A.; Lee, C.; Joseph, P.; Marchini, S.; Baccarini, A.; Kolev, V.; Romualdi, C.; Fruscio, R.; Shah, H.; Wang, F.; et al. MicroRNA-181a Has a Critical Role in Ovarian Cancer Progression through the Regulation of the Epithelial-Mesenchymal Transition. Nat. Commun. 2014, 5, 2977.
- Milenkovic, D.; Deval, C.; Gouranton, E.; Landrier, J.F.; Scalbert, A.; Morand, C.; Mazur, A. Modulation of MiRNA Expression by Dietary Polyphenols in ApoE Deficient Mice: A New Mechanism of the Action of Polyphenols. PLoS ONE 2012, 7, e29837.
- Hui, C.; Yujie, F.; Lijia, Y.; Long, Y.; Hongxia, X.; Yong, Z.; Jundong, Z.; Qianyong, Z.; Mantian, M. MicroRNA-34a and MicroRNA-21 Play Roles in the Chemopreventive Effects of 3,6-Dihydroxyflavone on 1-Methyl-1-Nitrosourea-Induced Breast Carcinogenesis. Breast Cancer Res. 2012, 14, R80.
- Ohno, M.; Shibata, C.; Kishikawa, T.; Yoshikawa, T.; Takata, A.; Kojima, K.; Akanuma, M.; Kang, Y.J.; Yoshida, H.; Otsuka, M.; et al. The Flavonoid Apigenin Improves Glucose Tolerance through Inhibition of MicroRNA Maturation in MiRNA103 Transgenic Mice. Sci. Rep. 2013, 3, 2553.
- Zhou, H.; Chen, J.X.; Yang, C.S.; Yang, M.Q.; Deng, Y.; Wang, H. Gene Regulation Mediated by MicroRNAs in Response to Green Tea Polyphenol EGCG in Mouse Lung Cancer. BMC Genom. 2014, 15, S3.
- Yamashita, S.; Lin, I.; Oka, C.; Kumazoe, M.; Komatsu, S.; Murata, M.; Kamachi, S.; Tachibana, H. Soy Isoflavone Metabolite Equol Inhibits Cancer Cell Proliferation in a PAP Associated Domain Containing 5-Dependent and an Estrogen Receptor-Independent Manner. J. Nutr. Biochem. 2022, 100, 108910.
- Murata, M.; Nonaka, H.; Komatsu, S.; Goto, M.; Morozumi, M.; Yamada, S.; Lin, I.C.; Yamashita, S.; Tachibana, H. Delphinidin Prevents Muscle Atrophy and Upregulates MIR-23a Expression. J. Agric. Food Chem. 2017, 65, 45–50.
- Yamada, S.; Tsukamoto, S.; Huang, Y.; Makio, A.; Kumazoe, M.; Yamashita, S.; Tachibana, H. Epigallocatechin-3-O-Gallate up-Regulates MicroRNA-Let-7b Expression by Activating 67-KDa Laminin Receptor Signaling in Melanoma Cells. Sci. Rep. 2016, 6, 19225.
More