Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Yi Wang and Version 2 by Catherine Yang.
The biological impact of ionizing radiation (IR) on humans depends not only on the physical properties and absorbed dose of radiation but also on the unique susceptibility of the exposed individual. A critical target of IR is DNA, and the DNA damage response is a safeguard mechanism for maintaining genomic integrity in response to the induced cellular stress. Unrepaired DNA lesions lead to various mutations, contributing to adverse health effects.
- radiosensitivity
- radioresistance
- genome editing
1. Gene Expression Analysis (Microarray and RNA-Sequencing)
The question-driven (hypothesis-generating) high-throughput genetic screening studies, set to explore the unknown in an unbiased manner, have been more prominent with the development of critical “omics”–related technologies such as RNA-sequencing, microarray, RNAi, and CRISPR-Cas9. The high-throughput screening can identify novel genes or regulators of specific phenotypes and generate novel hypotheses that can be validated in low-throughput mechanistic and functional studies (Figure 1).
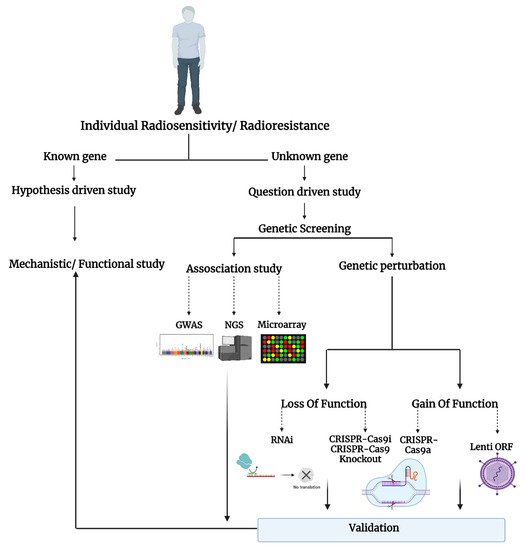
Figure 1. Schematic representation of the studies on individual radiosensitivity.
Using high-throughput gene expression methods, several studies have identified genes that influence the response to radiation. For instance, using DNA microarray analysis in lung cancer cells, Guo et al. profiled global gene expression in response to IR [1][20]. A microarray contains thousands of engineered complementary DNA (cDNA) oligonucleotides known as probes that hybridize with specific fluorescently labeled RNA molecules, and the expression of different known transcripts can be detected simultaneously [2][21]. Guo et al. focused their analyses on the expression of 143 genes in 2 lung cancer cell lines (NCI-H446 cells versus A549 cells) with different radiosensitivities in response to a single 5 Gy dose of gamma rays [1][20]. Compared to radiosensitive NCI-H446 cells, the expression of XRCC5, ERCC5, ERCC1, RAD9A, ERCC4, and MDM2, genes involved in DNA repair mechanism, was significantly increased in the radioresistant A549 cell line. The authors suggested this list of genes may prove useful in attempts to sensitize radioresistant lung neoplasms [3][22].
Performing next-generation RNA-sequencing (RNA-seq), extensive studies have been done on gene expression alterations in response to IR in a cell population as a whole. In a search for a predictor of response to IR in cancer cells, Young et al. took an RNA-seq approach to analyze the gene expression in radiosensitive LNCaP and radioresistant PC-3 prostate cancer cells [4][23]. They identified two canonical pathways with opposing responses in both cell lines 24 h after irradiation with high energy X rays: the DNA repair pathway (downregulation of BRCA1, RAD51, and FANCG in LNCaP and opposite pattern in PC-3 cell) and the cell cycle control of DNA replication pathway (downregulation of ORC1, CDC6 and the MCM genes with contrasting pattern in PC-3 cell). In another study, the global gene expression in human glioma cells was assayed after exposure to a dose of gamma-rays leading to growth arrest. It was revealed that the inactivation of proapoptotic signaling molecules and late activation of antiapoptotic genes might contribute to the radioresistance of gliomas [5][24]. Deep sequencing was utilized to delineate different layers in the transcriptional response to IR in human breast cancer cells. This study identified protein-coding and previously unidentified non-coding genes that were responsive to IR [6][25]. Thus, RNA-seq allows for the complete sequencing of the whole transcriptome while microarray only profiles predefined transcripts through probe hybridization. In RNA-seq, purified RNA from genes and gene variants (e.g., splicing isoforms) are sequenced directly (without the help of the probes) [2][21]. Therefore, whereas both microarray and RNA-seq can show large numbers of differentially expressed genes, RNA-seq reveals an unbiased screening of a broader range of gene expression with higher specificity and sensitivity, including novel, coding, and non-coding transcripts, compared to microarrays [7][26].
Bulk RNA-seq analysis described in the previous section conventionally measures transcripts in a mixture of cells which allows the measurement of only the average transcript expression in a cell population. Such traditional sequencing methods are unable to analyze a small number of cells found in rare populations and also lose cellular heterogeneity information. Single-cell RNA-sequencing (scRNA-seq) is an innovative NGS approach that has enabled the measurement of the whole transcriptome at a single-cell resolution and contributed to understanding changes in the transcriptional circuitry of individual cells within their natural microenvironment. A scRNA-seq method was used in two different studies to investigate the acquired radioresistance in esophageal squamous cell carcinoma cells (ESCC). These studies revealed the cellular heterogeneity and dynamic gene expression changes in irradiated ESCC cells along with the genes and signaling pathways related to the development of radioresistance [8][9][27,28]. Similarly, scRNA-seq of breast cancer cell line MDA-MB-231 with and without IR treatment using the barcoded Smart-seq2 technology revealed a heterogeneous cellular response to DNA damage induced by IR. scRNA-seq data analysis also identified potential biomarkers of radiation sensitivity including MCM3, MCM4 and SLBP genes involved in DNA replication [10][29]. Thus, single-cell sequencing technology has the power to delineate the heterogeneous response to IR in different cancer types and thereby improve treatment options.
2. Genome-Wide Association Study (GWAS)
GWAS examines variations that are presented in the form of single nucleotide mutation. When the frequency of these mutations is more than 1% of the population, they are called single nucleotide polymorphisms (SNPs) [11][33]. In one of the first clinical studies of SNPs, Kerns et al. used the GWAS method to investigate genetic variants associated with erectile dysfunction as an indicator of normal tissue damage experienced after radiation therapy (RT) in prostate cancer patients [12][34]. From the high-throughput analysis of 512,497 SNPs, rs2268363, which is located in a gene whose encoded product affects male gonad development and function (the FSHR gene), was strongly associated with the development of long-term side effects of RT. This strongly supports the feasibility of using the GWAS approach in exploring the association between genetic predisposition and radiation injury in normal cells [12][34].
Moreover, although radiation-induced germline mutations or heritable genetic diseases in children of irradiated parents are still not confirmed, strong evidence of the heritability of the radiosensitivity trait in human somatic cells has been established [13][14][35,36]. In an attempt to discover genes and SNPs that affect radiosensitivity, Zyla et al. used genomic analysis from human twin pairs with the GWAS method and showed that about 66% of CDKN1A (cyclin-dependent kinase inhibitor 1A) expression in response to radiation is heritable [15][37]. CDKN1A encodes protein p21, a downstream effector of p53, and is one of the key regulators in cell cycle regulation and arrest following DNA damage. CDKN1A abnormal expression is associated with acute sensitivity to radiation. Moreover, GWAS allowed identification of SNPs that are significantly associated with CDKN1A expression (i.e., rs205543 (ETV6 gene), rs2287505, and rs1263612 (KLF7 gene) are involved in CDKN1A transcription factors, rs6974232 (RPA3 gene), rs1133833 (AKIP1 gene), and rs17362588 (CCDC141 gene) are genes involved in DNA mismatch and RNA repair [15][37].
To construct a more precise and efficient polygenic risk model, Oh et al. used hundreds of SNPs and developed a machine learning algorithm called pre-conditioned random forest regression that signals even for small differential risks [16][47]. By applying this novel method to the GWAS cohort dataset of 368 prostate cancer patients treated with RT at a single institution, the team was able to identify the false positive SNPs and evaluated the importance of each SNP (the key biological function of each SNP) in inducing the radiotoxic outcomes [16][47]. However, the GWAS method comes with drawbacks that have been clearly discussed by Cano-Gamez et al. [17][31]. One major setback might be the lack of understanding of the roles of disease-associated loci in non-coding regions of the genome. As their role in gene expression regulation in different cell types or physiological contexts is still unclear, translating GWAS findings into clinical interventions might not be efficient [17][31]. Furthermore, the candidates found in GWAS or other methods discussed above need to be functionally validated. To achieve that, functional genomics techniques such as RNAi and CRISPR-Cas9 are powerful tools for analyzing gene function.
3. Genome-Wide RNAi Screening Method
RNA interference is a powerful method for loss-of-function genetic screens for key regulators and critical pathways involved in a particular phenotype [18][48]. This method has been used to knock down specific genes to investigate radiosensitivity of cancer cells [19][49]. For instance, using genome-wide RNAi screening to search for radioresistance genes in colorectal cancer cells (HCT116 and HCT15 cells), Wang et al. found that RFC4 knockdown significantly mitigates X-ray-induced DNA damage repair and enhances apoptosis [20][38]. The protein encoded by the RFC4 gene facilitates cellular DNA DSB repair via a non-homologous end joining (NHEJ)-mediated pathway in colorectal cancer cells, and therefore RFC4 upregulation is associated with tumor progression [20][38]. In addition, five more genes, including NCAPH (regulatory subunit of the condensin complex), SYNE3 (transmission of mechanical forces across the nuclear envelope and in nuclear movement and positioning), LDLRAD2 (receptor-mediated endocytosis), NHP2 (required for ribosome biogenesis and telomere maintenance), and FICD (ATP binding activity) were also identified as potential candidate radioresistance genes.
Herr et al. used the same method to find homologous recombination repair (HRR)-specific factors in response to IR [21][39]. Since an intact sister chromatid template would be used in the HRR process, this pathway offers more accurate and error-free repair for DSBs (in comparison to the NHEJ pathway) [22][50]. The authors identified CDC73, a protein encoded by the HRPT2 tumor suppressor gene, as a new regulator of HRR. By interacting with core histones of H2B and H3, CDC73 optimizes chromatin remodeling around DSBs and supports the accessibility of the DNA for downstream repair elements and events [21][39]. Van Haaften et al. exposed nematode Caenorhabditis elegans to 60 Gy of radiation and used a genome-wide RNAi technique to identify eight genes necessary to protect the germline against DNA DSB. Intriguingly, most of these newly identified genes with known human orthologs (i.e., Y65B4BR.4A (human: WWP2), H19NO7.2a (human: USP7, HAUSP), Y41C4a.10 (human: TCEB2), Y67D8C.5 (human: UREB1, LASU1), and C52D10.9 (human: SKP1A)) are expected to play a role in the targeted degradation of proteins via the ubiquitination function. RAD51, histones, CDC25A, and p53, all of which play a role in DSB response, are regulated by ubiquitination. This observation supports the idea that certain proteins activate or regulate the DSB response pathway by undergoing proteasomal activity [23][40]. Knockdown of these genes improved sensitivity to ionizing radiation and amplified chromosomal nondisjunction [23][40]. In another study, van Haaften et al. expanded their data by identifying more genes that are active agents in DNA damage response and RNA processing and trafficking that contribute to increased radiosensitivity of germ cells in C. elegans. In addition, the novel genes were found to be strongly conserved throughout animal evolution. Among genes with human homolog, ATM, ITGA6, NIPBL, NOB1, CAND1/TIP120, WWP2, and TopBP1 have been observed [24][41].
Although RNAi is a robust tool for genome-wide screening through the downregulation of gene expression at the mRNA level regardless of the target gene copy numbers, its off-target effects are also inevitable [25][51]. In fact, suppression of gene expression by RNAi might not be efficient, which may result in only a partial knockdown [25][51]. Many of these shortcomings of RNAi are effectively addressed by CRISPR-mediated gene editing technology.
4. Genome-Wide CRISPR-Cas9 Screening Method
Adopted from the bacterial immune system, Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas-associated protein 9, known as CRISPR-Cas9, is a novel technology that has revolutionized genome editing and gene therapy [26][52]. The CRISPR-Cas9 system comprises two biological components: the RNA-guided DNA endonuclease, Cas9, and the chimeric single-guide RNA (sgRNA) [27][28][53,54]. The sgRNA is loaded onto Cas9 and directed to a 20 bp region on the DNA target via base pairing. For functional gene editing, the target DNA must immediately precede a 5’ NGG sequence (N is any nucleotide), referred to as a protospacer adjacent motif (PAM). Cas9 promotes genome editing by inducing a DSB at the target genomic locus by re-direction to its target region. The cellular machinery then repairs the DNA DSB via NHEJ or HRR pathways [28][54].
This technique has been applied to investigate the effect of several genes (e.g., Hsp70, osteopontin, and HIF-1/2α) as critical regulators in radioresistance or radiosensitivity traits in different cell lines [29][30][31][55,56,57]. To develop a comprehensive approach and investigate radioresistance regulatory factors in the colorectal cancer (CRC) cell (RKO, HCT116, and SW620), Yu et al. applied genome-scale CRISPR sgRNA library in negative selection screens to identify radioresistance candidate genes. They found that DNA polymerase alpha 2 (POLA2), radical S-adenosyl methionine domain containing 2 (RSAD2), and microRNA5197-5p (miR-5197) had the most significant fold changes after IR exposure [32][45]. However, further investigation showed that overexpression of miR-5197 impaired radioresistance to a more considerable extent compared to other gene candidates. By inhibiting the expression of cell cycle regulatory protein CDK6 and promoting cell cycle arrest in the G1/S phase, miR-5197 contributes to IR-induced apoptosis in CRC [32][45]. The authors, however, emphasized the need for further studies with an in vivo model to prove their findings [32][45]. Using a genome-wide CRISPR-Cas9 sgRNA library for the first time in nasopharyngeal carcinoma (NPC) cells and performing high-throughput sequencing on sgRNAs obtained in a negative screen, Ziyan et al. found nine genes involved in the radiosensitivity or radioresistance of NPC cells [33][43]. Five genes (BLN5, FAM3C, MUS81, DNAJC17, and CALD1) were suggested as radiosensitivity modulators, whereas four genes (CDKN2AIP, SP1, TOMM20, and SNX22) seemed to be potentially radioresistant genes. Additionally, an enrichment analysis of the KEGG database showed that these genes contribute to radiosensitivity or radioresistance in NPC via the Fanconi anemia pathway and TGF-beta signaling pathway. Through CRISPR/Cas9 high-throughput screening and negative selection of crucial genes that might be linked to radioresistance in NPC, Shen et al. also demonstrated that overexpression of LUC7L2 contributes to radioresistance via the autophagy process. LUC7L2 is an RNA binding protein that has not been fully studied and only has been characterized in recent years [34][58].
Hayman et al. performed a whole-genome CRISPR-Cas9 screen in an HNSCC cell line using treatment with ionizing radiation as a positive selection pressure to identify regulators of radiation sensitivity. Positive screening and NGS of sgRNAs enriched after multiple rounds of irradiation showed that activation of stimulator or interferon genes (known as STING, a signaling molecule associated with the endoplasmic reticulum) influences radiation response in HNSCC cells [35][44]. They further show that pharmacological activation of STING enhances the effects of ionizing radiation in vivo and might be a promising approach to enhance radiotherapeutic response in patients suffering from HNSCC. In an interesting study, Zhu et al. performed genome-wide CRISPR activation screening and identified calcium-regulated heat-stable protein 1 (CARHSP1) as an essential element involved in radioresistance traits in human glioblastoma cells [36][42]. Because of its cold-shock domain, CARHSP1 has the capacity to bind to polypyrimidine regions of single-stranded RNA, single-stranded DNA, or double-stranded DNA [37][59]. Hence, CARHSP1 can bind to DNA and regulate the rate of transcription termination, but also it has the potential to regulate RNA stability, mRNA degradation, and ribosomal translation [38][60]. Intriguingly, CARHSP1 enhances mRNA stability of tumor necrosis factor-alpha (TNF-α), a crucial pleiotropic cytokine and a critical inflammatory molecule [38][60]. With this information, Zhu et al. showed that an elevated level of CARHSP1 is associated with radioresistance of glioblastoma cells via CARHSP1/TNF-α pathway signaling [36][42]. Cheng et al. used an unbiased genome-wide CRISPR/Cas9 knockout strategy in A549 lung cancer cells and identified plakophilin 2 (PKP2) as a critical driver of radiation resistance in lung cancer cells [39][61]. Cheng et al. have shown for the first time that methylated PKP2 protein promotes NHEJ and increases lung cancer radioresistance. Arginine methylation of PKP2 is mediated by protein arginine methyltransferase-1 (PRMT1). Hence, PRMT1 inhibition may also be an attractive approach to radiosensitize lung cancer [39][61].