Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Johannes Tobias Thiel and Version 2 by Peter Tang.
Soft tissue sarcomas (STSs) are rare malignant conditions with more than 70 subtypes that are difficult to treat, especially in advanced or metastatic states. Next-generation sequencing technologies have provided comprehensive information and developed personalized medicine for treating cancer in general and STSs in particular. Growing knowledge of diverse gene alterations and biomolecular targets in various subtypes of STSs raises hope for novel treatment approaches and heralds a paradigm shift in the treatment of STSs. Activated cyclin-dependent kinases (CDKs) appear to play a critical role in sarcoma development and represent important targets for sarcoma therapy.
- CDK
- cyclin-dependent kinase
- sarcoma
- soft tissue sarcoma
1. Introduction
Soft tissue sarcomas (STSs) are rare, heterogeneous malignant tumors that accounts for about 1–2% of all cancers. In the United States, there are 12,750 new cases diagnosed yearly, and STSs kill 5270 people each year [1]. The crude incidence rate of STSs is 4.71 per 100,000 people in Europe, with an estimated 25,851 new cases in the European Union [2]. Soft tissue sarcoma is currently composed of approximately 80 subtypes defined by the World Health Organization (WHO), classified based on a combination of unique morphological, immunohistochemical, and molecular characteristics [3]. Although the ultimate cellular origin of sarcoma subtypes remains unclear, there is increasing evidence that they arise de novo from mesenchymal pluripotent stem cells [4][5][4,5].
The mainstay of therapy has been surgical resection with negative margins, but the prognostic impact of tumor-free margins on prognosis remains controversial [6][7][6,7]. The risk of recurrence and distant metastasis (DM) is mainly related to tumor biology. There are significant variations in the incidence of DM across different sarcoma histologies, and the tumor grade and size impact this risk significantly in STSs [8]. Overall, the estimated five-year survival for STSs is ~57–62% and can vary widely depending on the disease stage and the complex interplay between the anatomical site and STS subtype [9]. Patients with advanced STSs have a median overall survival of fewer than 18 months and require systemic therapies, which unfortunately have not been very promising so far [10][11][12][10,11,12].
Additionally, due to the fact that STSs are heterogeneous, responses to generalized therapy and active substances are variable and usually no longer translate among unique subtypes [13]. Therefore, the treatment for each sarcoma subtype should be individual and personalized. To accomplish this goal, biomarkers and critical points in the signaling pathways for growth and progression must be elucidated and characterized. Recent advances advise that changes in cyclin-dependent kinase (CDK) pathways are vital drivers of sarcomagenesis specifically and of cancer in general [9][10][14][15][9,10,14,15].
2. What Are Cyclin-Dependent Kinases (CDKs)?
Cyclin-dependent kinases (CDKs) were first discovered through genetic and biochemical studies in various organisms, including yeasts and complex organisms such as frogs. With their discovery came a growing understanding of the importance of CDKs in cell reproduction [16][17][16,17]. In the 1960s, the cell-cycle phase in eukaryotic cells was described as a sequence of four phases (see Figure 1). A few years later, in 1987, the first CDK was described, cell division cycle 2 (cdc2), again changing the understanding of cell-cycle progression. As scientists discovered cdc2 first, it was named CDK1 [18]. CDKs are serine-threonine kinases; they phosphorylate their substrates at serines and threonines. The enzymes regulate transcription and mRNA processing and may also be involved in neuronal differentiation [10]. CDKs have no function in resting cells because of a structural confirmation that obscures the catalytic and substrate-binding domains [10][19][20][10,19,20]. Their serine/threonine-specific catalytic core partners, called cyclins, inherit regulatory subunits, controlling kinase activity and substrate specificity [19]. Specific subsets of cyclins and CDKs regulate each phase transition in the cell cycle. Therefore, CDKs are essential enzymes that control the transition of the individual phases in the cell cycle through restriction points in a compassionate manner [21].
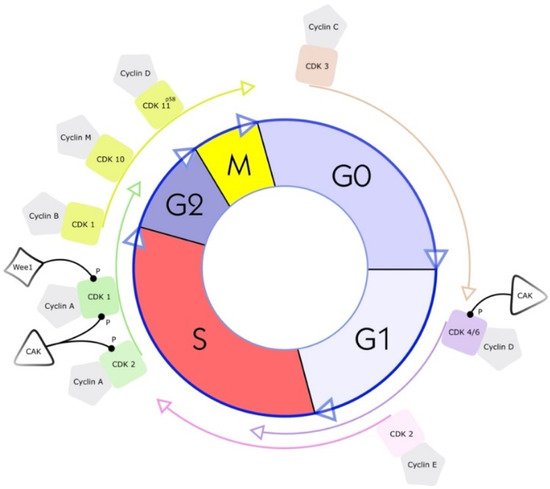
Figure 1. Cell cycle and a simplified illustration of interactions with selected kinases and cyclins. G1 is the cell cycle phase; in this phase, the cell increases in volume, but mitosis has not yet taken place. It is the first part of the interphase and transition into the S phase, the cell cycle’s replication phase. DNA replication takes place in this phase. The S phase (2nd phase) is usually constant in time (about 7 to 8 h) and lies between the G1 and G2 phases. Third, the G2 phase of the cell cycle is the second part of the interphase. It follows the S phase and enters the prophase of mitosis, in which the cell’s chromatin condenses into chromosomes. The following M phase describes the division of the cell. The nucleus splits (mitosis), and the entire cell divides (cytokinesis). Finally, the facultative G0 phase is the cell-cycle stage in which dormant or differentiated cells (e.g., nerve and muscle cells) are found [18].
CDK–cyclin complex activity is tightly regulated by many CDK inhibitors (CKIs), which stop the cell-cycle progression under unfavorable conditions [22]. To date, 20 different CDKs (numbered from CDK1 to CDK20) and 29 human cyclins and cyclin-like proteins have been identified [18]. UniProtKB IDs list the functions, structures, sequences, and interactions of many known CDKs, accessible at the website https://www.uniprot.org/uniprot/ (accessed on 16 May 2022) [20].
CDKs were traditionally divided into two groups: CDKs of the first group can bind multiple cyclins and regulate the cell cycle progression (CDKs 1–4, 6 and 7). CDKs of the second group form complexes with a single cyclin and are involved in regulating transcription processes (CDKs 7–9,12, 13 and 19) [23][24][25][26][23,24,25,26]. CDKs 5, 10, 11, 14–18, and 20 do not fit into the abovementioned categories. They lack explicit functional annotations and have different functionalities, which are often tissue specific [23]. CDK5, for example, cannot directly control cell-cycle regulation [27]. It regulates neuronal development and post-mitotic neuronal activities by binding with p35 [28]. Substrates of CDK5, such as transcription factor p53 and myocyte enhancer factor 2 (MEF2), are involved in sarcoma progression [29][30][29,30].
There is increasing evidence that the impaired activation and expression of CDKs are associated with tumors; conversely, targeting CDKs in tumor cells has become a promising therapeutic strategy [32]. The inhibition of CDKs can reduce the growth and progression of sarcoma cells and lead the diseased cells into apoptosis [20][32][20,32].
3. Selected CDKs and Their Role in Sarcoma Research and Treatment
CDKs are focused on providing targeted therapy to patients suffering from sarcoma. However, the same treatment differs in efficacy in different patients and tumors. These results reflect the unique microenvironment of each tumor. In addition to the specific microenvironment, compensatory pathways that undermine the mechanism of new CDK-related treatments have also been discovered [9][33][9,33]. Based on sequence homology, scientists have mapped and grouped CDKs, cyclins, and CKIs. As more and more information has been collected, it has become clear that the earlier, rather strict criteria for classifying these proteins are no longer correct. Recently, studies have shown that complexes of CDK and cyclin subunits are themselves highly active [19].
3.1. CDK1
CDK1 (CDC2) plays a vital role during the cell cycle. This enzyme strongly regulates the S phase and the G2 phase. The separate binding of cyclin A and cyclin B to CDK1 drives the transition from the G2 phase to the M phase [32][34][32,34]. Experiments with knockout CDK1 mice have shown that CDK1 is essential for initiating mitosis [34]. The phosphorylation of the complex of CDK1 and cyclin B by Wee1, a serine/threonine kinase, leads to the inhibition of CDK1 (see Figure 1) [32]. Therefore, by inhibiting the inhibitor Wee1, the activity of CDK1 can be increased. The proof of this principle has been shown using the CKI MK1775 (adavosertib), a Wee1 inhibitor in different cell lines, derived from human liposarcomas (LPSs) and from rhabdomyosarcomas (RMSs). Notably, in these cell lines, CDK1 is strongly expressed during the progression of the S phase and the transition from the G phase to the M phase. The proliferation ability of the cells is decreased by the inhibition of CDK1 expression or activation [35][36][35,36].
3.2. CDK2
CDK2, similar to CDK1, is a serine/threonine kinase and is involved in the transition from the G1 phase to the S phase and is closely associated with cyclins A and E (see Figure 1). For the treatment of STSs, p27, a tumor suppressor protein, is of interest. P27 inhibits CDK2. Therefore, upregulation of p27 in a human RMS cell line results in potent inhibition of CDK2, and decreases the proliferation ability of cells [37][38][37,38]. Transforming growth factor-beta (TGFβ1) was found to initiate the upregulation of p27 in RMS. It also enhances the binding affinity of p27 to the complex of CDK2 and cyclin E [37]. Due to these two mechanisms, TGFβ1 is a promising inhibitor of tumor cell proliferation in RMS, round liposarcomas, and myxoid cell lines. The cell line HS-18 derived from human LPSs highly expresses CDK2 and cyclin A and cyclin E [39][40][39,40]. This high expression of CDK2 and genetic aberrations in the coding sequences for CDK2 in sarcoma have also been associated with a bad clinical course. Therefore, CDK2 gene aberrations are considered crucial prognoses influencing factors [41].
3.3. CDK4 and CDK6
CDK4 and CDK6 both interact with D-type cyclins. Three D-type cyclins are currently known: cyclins D1, D2, and D3. Not only are binding partners identical, but also 71% of the amino acid identity in CDK4 and CDK6 is the same [32]. The crucial role of both proteins is to promote the progression of the G1 phase and the transition from the G1 phase to the S phase (see Figure 1).
P16 (also known as CDK inhibitor 2A) and p21 (also known as CDK inhibitor 1A) are familiar CKIs for CDK4 and CDK6; both p proteins are tumor suppressors. CDK4 and CDK6 can phosphorylate the retinoblastoma tumor suppressor protein (Rb1). In this way, both CDKs inactivate Rb1 and silence multiple genes [42].
The interplay of Rb1, CDK4, and CDK6 influences cancer cell proliferation, differentiation, and transformation [43]. In sarcomas, research on CDK4 and CDK6 as well as CKIs such as palbociclib is still ongoing and the most promising, even if the majority of studies are experimental. Palbociclib is a potent inhibitor (CKI) of both CDK4 and CDK6. Rb1-proficient ovarian cancer cell lines are sensitive to palbociclib; in contrast, glioblastoma multiforme cell lines are highly resistant to palbociclib [44][45][44,45]. These cell lines inherit deletions or mutations in the Rb1 gene. This highlights that active, hypophosphorylated Rb1 is key to the efficacy of palbociclib. By confronting Rb1-deficient cell lines with extremely high levels of CKI, the principle was proven. Even high concentrations of palbociclib have failed to induce G1 arrest [45]. Some authors have suggested that Rb1 is a predictive biomarker for response to CDK4 and CDK6 inhibitors [32].
A total of 85% of myxoid and round cell liposarcoma highly express both CDKs. Rb1 immunoreactivity has been reported in 66% of these sarcoma subtypes [46]. Scientists have observed a significant overexpression of CDK4 and CDK6 in mouse model, linked with the progression and occurrence of both dedifferentiated (DDLPSs) and well-differentiated liposarcomas (WDLPSs) [47][48][47,48].
CDK4 may even be used as a prognostic marker, as poor disease-specific survival was associated with high expression of CDK4 in 56 patients with LPSs [49]. Additionally, a significantly high expression of CDK4 has been found in patients with WDLPSs recurrence after surgery [49]. Complementary low expression of CDK4 in these tumors was associated with a better prognosis and a higher progression-free survival [50].
In addition to palbociclib, ribociclib is another interesting CKI, also known as LEE011, and a selective CDK4 inhibitor. This drug arrests liposarcoma tumor cells in the G0 phase and G1 phase after 24 h of incubation and limits tumor cell proliferation [51]. Few clinical trials have been designed with this drug, and the majority of registered clinical trials are still ongoing. Recently, a phase Ib study in patients with LPSs and ribociclib has been completed [52]. Together with other clinical trial results, these are discussed in Section 5. CDK4 and CDK6 are very similar from the ultrastructural point of view. Interestingly, the efficacy of dual CDK4/6 inhibitors against CDK4 and CDK6 is different. In vitro studies have found that the dual inhibitors abemaciclib, ribociclib, and trilaciclib were more powerfully inhibiting CDK4 than CDK6, while palbociclib, in contrast, had comparable efficacy against both CDK4 and CDK6 [53][54][55][53,54,55]. Finally, abemaciclib and trilaciclib not only inhibit CDK4/6 but also have a slight inhibitory effect on CDK5 and CDK9.
In RMS, CDK4/6 inhibitors appear to be able to arrest tumor growth. CDK4 knockdown mice receiving RMS cell lines showed impaired proliferation and poor transformation of tumor cells arrested in G1 phase. Deficient Rb1 phosphorylation induced this arrest [56]. In 2015, RMS expressing low levels of CDK4 were shown to be especially sensitive to ribociclib and, therefore, the inhibition of CDK4/6 [57].
In summary, there are few trials on CDK4 and CDK6 inhibitors in the treatment of STS, most of which are in the preclinical or early clinical stage [32][58][59][60][61][32,58,59,60,61].
3.4. CDK9
CDK9 is the catalytic subunit of two enzymes: one is Tat-activating kinase and the other is positive transcription elongation factor b (pTEFb) [61]. CDK9 is present and expressed during the whole cell cycle [62]. CDK9 supplies the transcriptional homeostasis and, therefore, fundamental regulation of gene transcription [63]. In malignancies, physiological homeostasis of transcription is generally absent; oncogenes take control and modulate transcription. Therefore, CDK9 is of great interest for targeted-therapy concepts because of its function as a “guardian of cellular transcriptional homeostasis” [20][32][64][65][20,32,64,65]. The interaction of CDK1/CDK2 and CDK9 can lead to apoptotic arrest in G2 and M phases causing inhibition of the whole cell cycle [66].
Alterations in CDK9 activity in RMS are suspected to inhibit the physiological differentiation of tumor cells [65][67][68][65,67,68]. Synovial sarcoma also shows a clear correlation between poor prognosis and high levels of CDK9 [69]. In chronic lymphocytic leukemia, small-cell lung cancer, and breast cancer, two CDK9 inhibitors (alvocidib (flavopiridol) and seliciclib) have already been applied [70][71][70,71].
3.5. CDK11
CDK11 differs from other CDKs; it is not only encoded by a single gene but by two genes. CDC2L1 (CDK11B) and CDC2L2 (CDK11A) share many bases homologously. CDC2L2 is specific to humans and is absent in mice [72][73][72,73]; both conventional groups of CDKs do not apply to CDK11 due to its variety of tasks [74]. In eukaryotic cells, a minimum of ten isoforms have been cloned already. The most common and highly active of them is CDK11p110 [75][76][77][75,76,77]. CDK11p110, similar to all CDK11 isoforms, is associated with RNA splicing, transcriptional regulation, and cell division [78]. Although very similar, the isoforms slightly vary in their way of functioning. CDK11p110 forms a complex with cyclin L and mainly interacts with RNA processing and transcription, whereas CDK11p58 pushes mitosis and acts kinase specific in the G2 and M phase [75][78][79][80][81][75,78,79,80,81].
LPS tissue microarrays analyzed by immunohistochemistry showed high levels of CDK11. Benign lipoma tissue, in contrast, expressed significantly less CDK11 [74]. First attempts with synthetic lentiviral shRNA and siRNA suppressing CDKs successfully induced and increased doxorubicin’s cytotoxic capability in LPS cells [74]. However, CDK 11 isotypes’ functions cannot be simplified. Scientists have proven the antagonistic effects of CDK11p58 and CDK11p110 in breast and prostate cancer. CDK11p58 appears to induce anti-metastatic and anti-proliferation effects. In contrast, CDK11p110 promotes the cell viability and survival abilities of cell clones [82][83][84][82,83,84].
Although research has intensified on CDK11 inhibitors, no single CKI has been designed yet.