Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Spyros Zissimopoulos.
The ryanodine receptor (RyR2) has a critical role in controlling Ca
2+
release from the sarcoplasmic reticulum (SR) throughout the cardiac cycle. RyR2 protein has multiple functional domains with specific roles, and four of these RyR2 protomers are required to form the quaternary structure that comprises the functional channel. Numerous mutations in the gene encoding RyR2 protein have been identified and many are linked to a wide spectrum of arrhythmic heart disease. Gain of function mutations result in a hyperactive channel that causes excessive spontaneous SR Ca
2+
release, but rare hypoactive loss of function mutations have also been described.
- ryanodine receptor
- catecholaminergic polymorphic ventricular tachycardia
- calcium release deficiency syndrome
1. Introduction
Cardiac contraction is initiated by a wave of electrical activity (action potential) triggering a rapid rise in intracellular Ca2+ concentration, which activates sarcomere contraction, followed by repolarization and Ca2+ removal that allows the heart to relax and refill [1]. Gene mutations in any of the proteins involved in excitation–contraction coupling (ECC) can lead to arrhythmogenic cardiac disease, most commonly, long QT and Brugada syndromes (repolarization abnormalities), catecholaminergic polymorphic ventricular tachycardia (CPVT) (dysregulated intracellular Ca2+), and dilated and hypertrophic cardiomyopathies (sarcomere dysfunction) [2]. Cardiomyocyte Ca2+ homeostasis is indeed an arrhythmogenic substrate [3]. For contraction of the adult human heart, plasma membrane depolarization induces Ca2+ influx via voltage-gated Ca2+ channels (also known as L-type Ca2+ channel or dihydropyridine receptor) located on invaginations of the sarcolemma called transverse (T)-tubules. This by itself is insufficient to trigger sarcomere contraction, but the rapid rise in Ca2+ activates ryanodine receptors (RyR2) located on the immediately opposing junctional sarcoplasmic reticulum (SR) through which a much larger Ca2+ release occurs in the form of Ca2+ sparks [4]. The near-synchronous activation of ~104 Ca2+ sparks at the start of the action potential produces the cytosolic Ca2+ transient that activates the contractile apparatus within cardiac myocytes. For relaxation, cytosolic Ca2+ concentration returns to baseline with Ca2+ extrusion outside of the cell via the Na+/Ca2+ exchanger (NCX) to remove an amount equal to the extracellular Ca2+ entry source, and Ca2+ uptake into the SR via the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) to remove an amount equal to the SR Ca2+ release source. Imbalances in cardiomyocyte Ca2+ cycling, namely the Ca2+ fluxes across the plasma and SR membrane, can alter the action potential and lead to arrhythmia.
2. RyR2 Structure-–Function Relationships
The RyR is the largest known ion channel, consisting of four identical subunits of ~5000 amino acids each, which combine to form a single Ca2+ permeable pore. Three RyR isoforms are expressed in mammals, RyR1 (the predominant isotype in skeletal muscle), RyR2 (the predominant isotype in cardiac muscle), and RyR3. Considering its size, complexity, and responsibility for achieving both short-(beat-to-beat) and long-term maintenance of heart contraction, it is not surprising that many factors fine-tune RyR2 function. In the following, wresearchers aim to describe determinants of RyR2 regulation as interacting layers consisting of: the pore-forming domains, domains that directly interact with the pore, domains that indirectly regulate the pore through intermediaries, or interacting regulatory proteins. Recent high-resolution cryo-electron microscopy (EM) studies reveal that the long RyR polypeptide chain is organized in distinct structural domains that are predominantly α-helical and face the cytoplasm [9,10][5][6] (Figure 1). They include the N-terminus domain (NTD) composed of sub-domains A/B/C, three SPRY domains including RyR repeat (RY) 1&2, junctional solenoid (JSol), bridging solenoid (BSol) consisting of sub-domains B&C flanking RY3&4, and core solenoid (CSol). According to a different nomenclature by Peng et al. [10][6], JSol is known as the “handle domain”, BSol is known as the “helical domain” but it was previously referred to as the “central domain” [11][7], and CSol is also known as the “central domain”. To avoid confusion with “central domain” terminology, wresearchers adopt the nomenclature by des Georges et al. [9][5]. The C-terminal ~500 residues contain the transmembrane domain (TMD) composed of six segments that anchor the protein on the SR membrane, followed by the short cytosolic C-terminus domain (CTD). The pore is formed by the S6 lining helix supported by S5 and the S5-S6 loop containing the selectivity filter.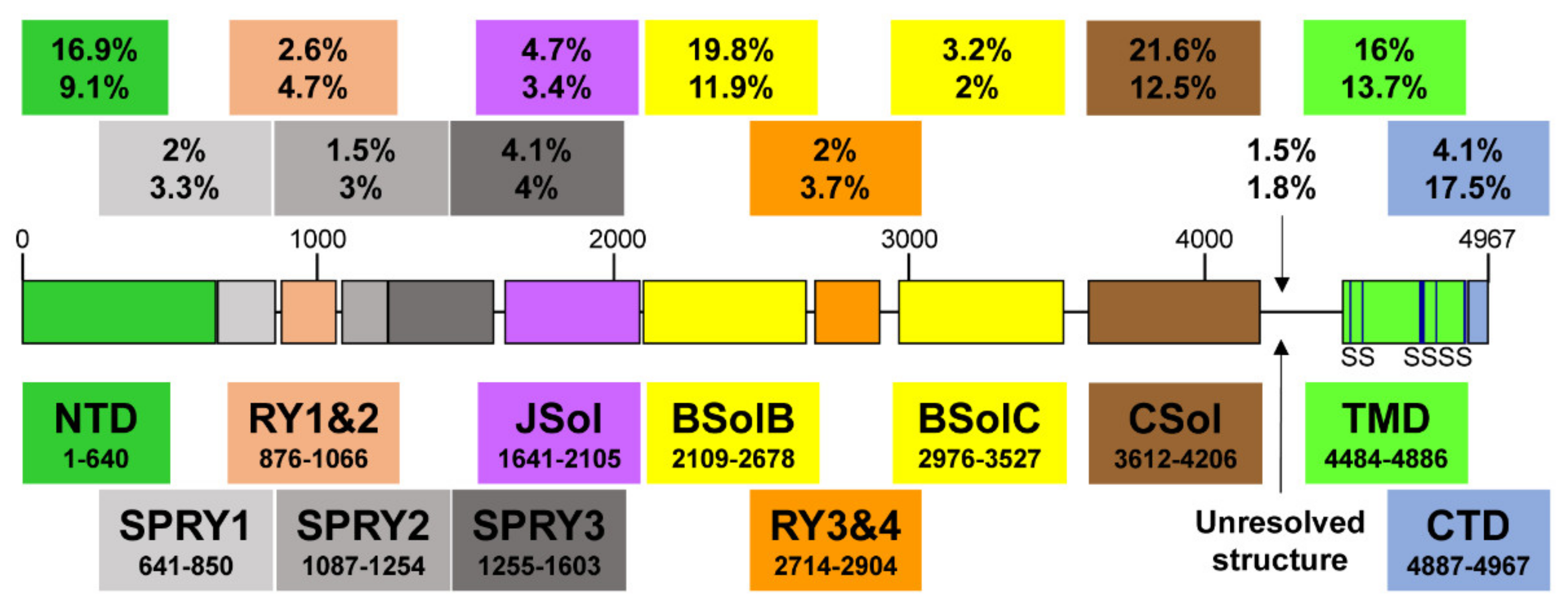
Figure 1. A schematic illustration of human RyR2 domain organization and boundaries. Color-coded distribution of mutations associated with disease (top value) and their frequency within individual structural domains (bottom value). The six transmembrane segments (S) are represented by a vertical line. See Figure 2 below for a corresponding cartoon representation showing the arrangement of domains in 3D (using same color coding).
RyR2 Channel Self-Assembly
Although RyR2 is a macromolecular complex with cytosolic, SR integral, and luminal accessory proteins, the 4-subunit RyR2 self-assembly is necessary and sufficient to form a fully functional Ca2+ release channel. To understand how RyR2 is auto-regulated, wresearchers consider a reductionist, “bottom-up” approach starting from the pore. Expression of the last two TM segments together with the CTD forms a tetrameric cation permeable pore that is constitutively open [12][8]. The CTD self-associates to support RyR tetramerization required for the formation of functional channels [13,14][9][10]. On top of the pore sits the CSol acting as the channels’ gatekeeper. The CSol makes extensive contacts with the pore-forming region including the S2S3 loop, the extended cytosolic part of S6, and the CTD [9,10][5][6]. However, the expression of large constructs encompassing the CSol, TMD, and CTD form ion channels that remain largely open and leaky [15,16,17][11][12][13]. Clearly, the remaining domains are needed to support the closure of the pore and fully regulate the channel. They are not making direct contact with the pore-forming region, but their effects are mediated via the CSol. Indeed, the NTD, Jsol, Bsol, and CSol are involved in multiple intra- and inter-subunit interactions with each other [9,10,18,19,20][5][6][14][15][16]. The NTD also interacts with itself to promote channel closure as well as to support the formation of functional tetrameric RyR2 channels [21,22,23][17][18][19]. RyR2 activity is fine-tuned by the peripheral SPRY and RY domains, and their interactions with JSol and BSol [9,10][5][6].3. Physiological RyR2 Regulators
The principal physiological RyR2 regulator is Ca2+ acting from both the cytosolic and SR luminal side. RyR2 is mostly inactive at low diastolic (~100 nM) cytosolic Ca2+ concentration ([Ca2+]). Membrane depolarization causes a rapid rise in [Ca2+] from nanomolar to micromolar levels in the narrow dyadic cleft formed between T-tubule and SR junctional membrane, which increases RyR2 activity in a sigmoidal function of cytosolic Ca2+ concentration. RyR2 can be fully activated at ~100 μM Ca2+ (reviewed in [24,25][20][21]). Recent cryo-EM and mutagenesis studies suggest that the cytosolic Ca2+-binding site is located at the CSol–CTD interface with residues E3848, E3922, and T4931 (coordinates for human RyR2) critical for Ca2+ interaction [9,26,27,28][5][22][23][24]. Interestingly, although the Ca2+-bound RyR structure reveals a contraction of the CSol that facilitates the dilation of the S6 bundle, the pore remains closed [9,26][5][22]. This could be due to the in vitro experimental conditions of cryo-EM. RyR2 is also regulated by SR luminal Ca2+ (reviewed in [24,25][20][21]). Although luminal Ca2+ can indirectly activate RyR2 by acting on the cytosolic Ca2+-binding site following passage through the pore, a separate site accessible only to luminal Ca2+ is likely to exist. Mutagenesis analysis has indicated that residue E4872 within S6 is necessary for luminal Ca2+ activation without affecting cytosolic Ca2+ modulation [29][25].3.1. Regulation of RyR2 by Accessory Proteins
RyR2 is also regulated by accessory proteins and post-translational modifications (phosphorylation and oxidation) (reviewed in [25,30][21][26]). Here, wresearchers focus on three RyR2-associated proteins, FK506-binding protein (FKBP), calmodulin (CaM), and calsequestrin (CSQ2) because of their implication in genetic cardiac disease.3.1.1. FKBP12/12.6
In native cardiac tissue, both of the two FKBP homologues of 12 and 12.6 kDa are found associated with RyR2 depending on the animal species [31][27]. RyR2 affinity is much greater for FKBP12.6 than FKBP12; however, native RyR2 occupancy by FKBP12.6 is minimal due to the latter being expressed at very low levels [31,32,33][27][28][29]. Functional studies generally suggest that FKBP12.6 promotes RyR2 channel closure and prevents SR Ca2+ leak (reviewed in [34][30]). Cryo-EM RyR structures in complex with FKBP12/12.6 indicate that the binding site is composed of multiple determinants contributed by the SPRY1, SPRY3, NTD, and JSol domains [26,35,36][22][31][32]. FKBP12.6 stabilizes RyR2 in a closed state by inducing the relaxation of the CSol and strengthening the NTD-NTD inter-subunit interaction [26][22].3.1.2. Calmodulin (CaM)
CaM is a high-affinity, Ca2+-binding, dumbbell-shaped protein containing four EF-hand motifs, two of each on both N and C lobes. Both the Ca2+ free (apo-CaM) and Ca2+ bound forms of CaM interact with RyR2 to inhibit channel activity, although inhibition is more pronounced at high Ca2+ [37,38][33][34]. The cryo-EM RyR2 structures in complex with apo-CaM and Ca2+-CaM indicate that they bind to distinct sites [39][35]. The N-lobe of apo-CaM binds BSolB, whereas its C-lobe binds at the interface between JSol and CSol. Ca2+-CaM shifts position and interacts only with the CSol. Ca2+-CaM stabilizes RyR2 in a closed state by inducing conformational changes in the CSol to release the pulling force for the dilation of the S6 bundle. Notably, CaM mutations have been linked with arrhythmia and sudden cardiac death, with most cases diagnosed as long QT syndrome and CPVT (reviewed in [40,41][36][37]). Mutations in all three CALM genes, which produce identical CaM peptide sequences, have been reported. Most of the disease-associated CaM mutations studied to date seem to reduce CaM’s inhibitory action on RyR2.3.1.3. Calsequestrin-2 (CSQ2)
CSQ2 is a low-affinity, high-capacity Ca2+-binding protein of ~45 kDa located within the SR lumen, where it buffers the free Ca2+ concentration to ~1 mM. It is associated with RyR2 via the SR integral proteins triadin and junctin, as well as directly interacting with the S1S2 luminal loop of RyR2 [42,43][38][39]. It is generally believed that CSQ2 exerts an inhibitory effect on RyR2 Ca2+ release (reviewed in [44][40]). CSQ2 knockout mouse models present with stress-induced arrhythmia indicating that CSQ2 plays an important role in the cardiac function [45,46][41][42]. Indeed, CASQ2 is the second most mutated gene in CPVT accounting for ~5% of cases [41][37]. Initially described as the recessive form of CPVT (CPVT2), autosomal dominant mutations have also been reported [47,48,49][43][44][45]. CPVT-linked CSQ2 mutations may result in impaired Ca2+ buffering, multimer formation and/or RyR2 regulation [41,44,50][37][40][46]. To summarize, wresearchers propose a 4-layer model for cytosolic regulation of the RyR2 pore (Figure 2). The first layer directly blanketing the pore consists of the CSol. The second layer consisting of NTD, Jsol, and BSol transmits stimulatory/inhibitory signals to the pore via the CSol. The third layer consists of the SPRY and RY domains acting on the second layer. The final fourth layer consists of cytoplasmic ligands, namely accessory proteins and protein kinases. While small ligands such as Ca2+ may have access to the pore-forming region, the large size of proteins precludes them from doing so. The effects of cytoplasmic proteins feed into layers 1/2/3 to alter their conformation and RyR2 inter-domain interactions. For example, FKBP12.6 binding (to SPRY1, SPRY3, NTD, and JSol domains) directly affects layers 2&3, which in turn transmit an allosteric signal to the pore via layer 1. The effect of phosphorylation (mediated by protein kinases) also feeds into layers 1/2/3. S2808 and S2814, whose phosphorylation is implicated in RyR2 physiology and disease are located within RY3&4. The luminal regulation of the RyR2 pore is simpler. The effects of luminal ligands can be direct to the pore (e.g., luminal Ca2+) or via the small RyR2 luminal loops.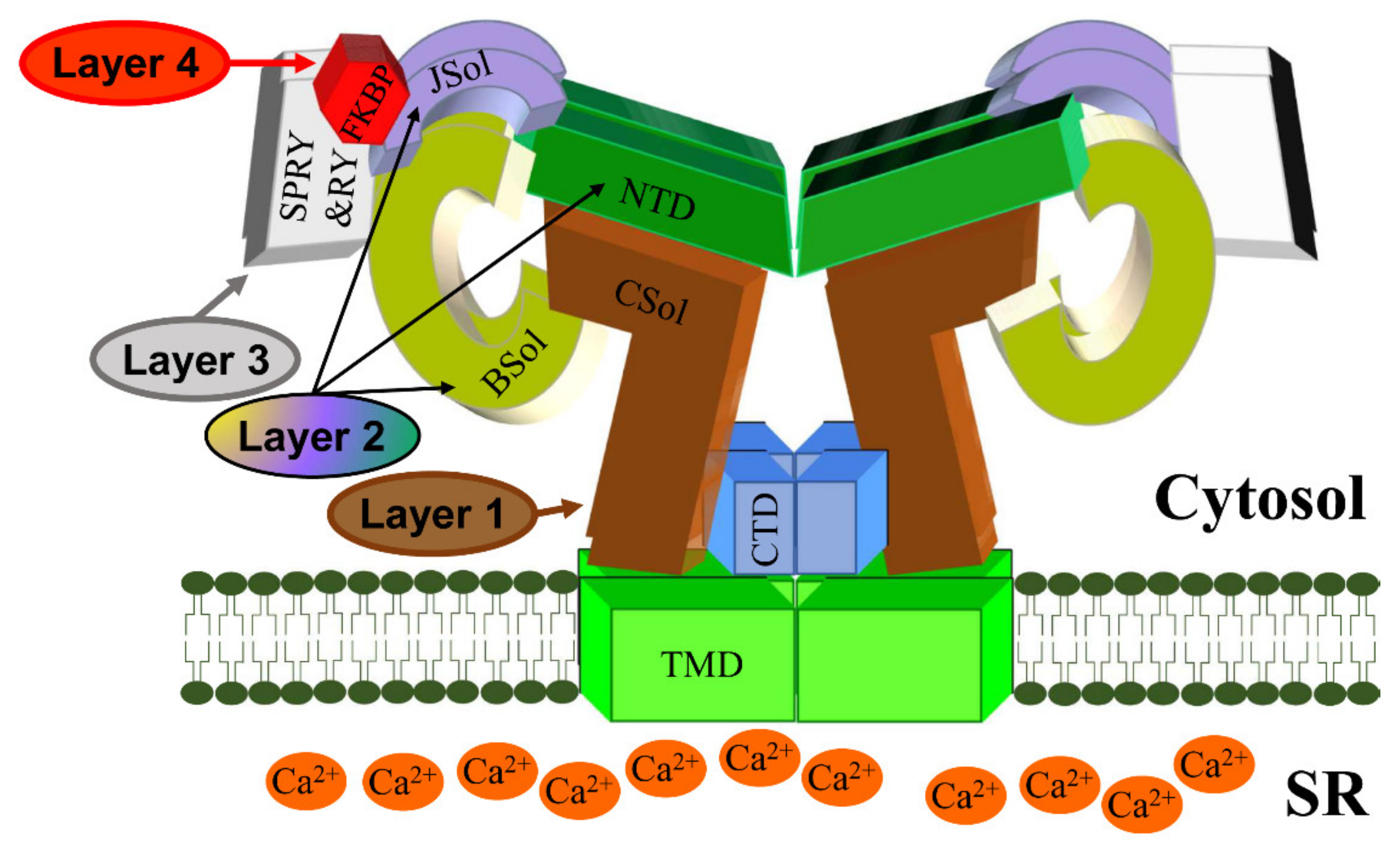
Figure 2. A schematic representation of the RyR2 3D architecture depicting 4 regulatory layers from the cytoplasmic side. The pore-forming region, comprised of the TMD and CTD, is modulated by the CSol (Layer 1), NTD, JSol and BSol (Layer 2), SPRY1/2/3 and RY1/2/3/4 (Layer 3), and accessory proteins (Layer 4), e.g., FKBP. The colors of regions in the RyR2 3D structure correspond to the colors in the RyR2 sequence in Figure 1.
References
- Bers, D. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205.
- Tester, D.J.; Ackerman, M.J. Cardiomyopathic and channelopathic causes of sudden unexplained death in infants and children. Annu. Rev. Med. 2009, 60, 69–84.
- Eisner, D.A.; Caldwell, J.L.; Kistamas, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195.
- Cheng, H.; Lederer, W.J.; Cannell, M.B. Calcium sparks: Elementary events underlying excitation-contraction coupling in heart muscle. Science 1993, 262, 740–744.
- Des Georges, A.; Clarke, O.B.; Zalk, R.; Yuan, Q.; Condon, K.J.; Grassucci, R.A.; Hendrickson, W.A.; Marks, A.R.; Frank, J. Structural Basis for Gating and Activation of RyR1. Cell 2016, 167, 145–157.e117.
- Peng, W.; Shen, H.; Wu, J.; Guo, W.; Pan, X.; Wang, R.; Chen, S.R.; Yan, N. Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science 2016, 354, aah5324.
- Yano, M.; Yamamoto, T.; Ikeda, Y.; Matsuzaki, M. Mechanisms of Disease: Ryanodine receptor defects in heart failure and fatal arrhythmia. Nat. Clin. Pract. Cardiovasc. Med. 2006, 3, 43–52.
- Euden, J.; Mason, S.A.; Williams, A.J. Functional characterization of the cardiac ryanodine receptor pore-forming region. PLoS ONE 2013, 8, e66542.
- Gao, L.; Tripathy, A.; Lu, X.; Meissner, G. Evidence for a role of C-terminal amino acid residues in skeletal muscle Ca2+ release channel (ryanodine receptor) function. FEBS Lett. 1997, 412, 223–226.
- Stewart, R.; Zissimopoulos, S.; Lai, F. Oligomerization of the cardiac ryanodine receptor C-terminal tail. Biochem. J. 2003, 376, 795–799.
- Bhat, M.; Zhao, J.; Takeshima, H.; Ma, J. Functional calcium release channel formed by the carboxyl-terminal portion of ryanodine receptor. Biophys. J. 1997, 73, 1329–1336.
- George, C.; Jundi, H.; Thomas, N.; Scoote, M.; Walters, N.; Williams, A.; Lai, F. Ryanodine receptor regulation by intramolecular interaction between cytoplasmic and transmembrane domains. Mol. Biol. Cell 2004, 15, 2627–2638.
- Xu, X.; Bhat, M.; Nishi, M.; Takeshima, H.; Ma, J. Molecular cloning of cDNA encoding a Drosophila ryanodine receptor and functional studies of the carboxyl-terminal calcium release channel. Biophys. J. 2000, 78, 1270–1281.
- Seidel, M.; de Meritens, C.R.; Johnson, L.; Parthimos, D.; Bannister, M.; Thomas, N.L.; Ozekhome-Mike, E.; Lai, F.A.; Zissimopoulos, S. Identification of an amino-terminus determinant critical for ryanodine receptor/Ca2+ release channel function. Cardiovasc. Res. 2021, 117, 780–791.
- Suetomi, T.; Yano, M.; Uchinoumi, H.; Fukuda, M.; Hino, A.; Ono, M.; Xu, X.; Tateishi, H.; Okuda, S.; Doi, M.; et al. Mutation-linked defective interdomain interactions within ryanodine receptor cause aberrant Ca2+ release leading to catecholaminergic polymorphic ventricular tachycardia. Circulation 2011, 124, 682–694.
- Uchinoumi, H.; Yano, M.; Suetomi, T.; Ono, M.; Xu, X.; Tateishi, H.; Oda, T.; Okuda, S.; Doi, M.; Kobayashi, S.; et al. Catecholaminergic polymorphic ventricular tachycardia is caused by mutation-linked defective conformational regulation of the ryanodine receptor. Circ. Res. 2010, 106, 1413–1424.
- Kimlicka, L.; Lau, K.; Tung, C.C.; Van Petegem, F. Disease mutations in the ryanodine receptor N-terminal region couple to a mobile intersubunit interface. Nat. Commun. 2013, 4, 1506.
- Seidel, M.; Thomas, N.L.; Williams, A.J.; Lai, F.A.; Zissimopoulos, S. Dantrolene rescues aberrant N-terminus inter-subunit interactions in mutant pro-arrhythmic cardiac ryanodine receptors. Cardiovasc. Res. 2015, 105, 118–128.
- Zissimopoulos, S.; Viero, C.; Seidel, M.; Cumbes, B.; White, J.; Cheung, I.; Stewart, R.; Jeyakumar, L.H.; Fleischer, S.; Mukherjee, S.; et al. N-terminus oligomerization regulates the function of cardiac ryanodine receptors. J. Cell Sci. 2013, 126, 5042–5051.
- Laver, D.R. Regulation of the RyR channel gating by Ca2+ and Mg2+. Biophys. Rev. 2018, 10, 1087–1095.
- Meissner, G. The structural basis of ryanodine receptor ion channel function. J. Gen. Physiol. 2017, 149, 1065–1089.
- Chi, X.; Gong, D.; Ren, K.; Zhou, G.; Huang, G.; Lei, J.; Zhou, Q.; Yan, N. Molecular basis for allosteric regulation of the type 2 ryanodine receptor channel gating by key modulators. Proc. Natl. Acad. Sci. USA 2019, 116, 25575–25582.
- Murayama, T.; Ogawa, H.; Kurebayashi, N.; Ohno, S.; Horie, M.; Sakurai, T. A tryptophan residue in the caffeine-binding site of the ryanodine receptor regulates Ca2+ sensitivity. Commun. Biol. 2018, 1, 98.
- Xu, L.; Chirasani, V.R.; Carter, J.S.; Pasek, D.A.; Dokholyan, N.V.; Yamaguchi, N.; Meissner, G. Ca2+-mediated activation of the skeletal-muscle ryanodine receptor ion channel. J. Biol. Chem. 2018, 293, 19501–19509.
- Chen, W.; Wang, R.; Chen, B.; Zhong, X.; Kong, H.; Bai, Y.; Zhou, Q.; Xie, C.; Zhang, J.; Guo, A.; et al. The ryanodine receptor store-sensing gate controls Ca2+ waves and Ca2+-triggered arrhythmias. Nat. Med. 2014, 20, 184–192.
- Zissimopoulos, S.; Lai, F. Ryanodine receptor structure, function and pathophysiology. In Calcium: A Matter of Life or Death; Elsevier: Amsterdam, The Netherlands, 2007; Volume 41, pp. 287–342.
- Zissimopoulos, S.; Seifan, S.; Maxwell, C.; Williams, A.J.; Lai, F.A. Disparities in the association of the ryanodine receptor and the FK506-binding proteins in mammalian heart. J. Cell Sci. 2012, 125, 1759–1769.
- Guo, T.; Cornea, R.L.; Huke, S.; Camors, E.; Yang, Y.; Picht, E.; Fruen, B.R.; Bers, D.M. Kinetics of FKBP12.6 binding to ryanodine receptors in permeabilized cardiac myocytes and effects on Ca sparks. Circ. Res. 2010, 106, 1743–1752.
- Jeyakumar, L.; Ballester, L.; Cheng, D.; McIntyre, J.; Chang, P.; Olivey, H.; Rollins-Smith, L.; Barnett, J.; Murray, K.; Xin, H.-B.; et al. FKBP binding characteristics of cardiac microsomes from diverse vertebrates. Biochem. Biophys. Res. Commun. 2001, 281, 979–986.
- MacMillan, D. FK506 binding proteins: Cellular regulators of intracellular Ca2+ signalling. Eur. J. Pharmacol. 2013, 700, 181–193.
- Yan, Z.; Bai, X.C.; Yan, C.; Wu, J.; Li, Z.; Xie, T.; Peng, W.; Yin, C.C.; Li, X.; Scheres, S.H.; et al. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature 2015, 517, 50–55.
- Zalk, R.; Clarke, O.B.; des Georges, A.; Grassucci, R.A.; Reiken, S.; Mancia, F.; Hendrickson, W.A.; Frank, J.; Marks, A.R. Structure of a mammalian ryanodine receptor. Nature 2015, 517, 44–49.
- Balshaw, D.; Xu, L.; Yamaguchi, N.; Pasek, D.; Meissner, G. Calmodulin binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor). J. Biol. Chem. 2001, 276, 20144–20153.
- Fruen, B.; Bardy, J.; Byrem, T.; Strasburg, G.; Louis, C. Differential Ca2+ sensitivity of skeletal and cardiac muscle ryanodine receptors in the presence of calmodulin. Am. J. Physiol. 2000, 279, C724–C733.
- Gong, D.; Chi, X.; Wei, J.; Zhou, G.; Huang, G.; Zhang, L.; Wang, R.; Lei, J.; Chen, S.R.W.; Yan, N. Modulation of cardiac ryanodine receptor 2 by calmodulin. Nature 2019, 572, 347–351.
- Chazin, W.J.; Johnson, C.N. Calmodulin Mutations Associated with Heart Arrhythmia: A Status Report. Int. J. Mol. Sci. 2020, 21, 1418.
- Priori, S.G.; Mazzanti, A.; Santiago, D.J.; Kukavica, D.; Trancuccio, A.; Kovacic, J.C. Precision Medicine in Catecholaminergic Polymorphic Ventricular Tachycardia: JACC Focus Seminar 5/5. J. Am. Coll. Cardiol. 2021, 77, 2592–2612.
- Handhle, A.; Ormonde, C.E.; Thomas, N.L.; Bralesford, C.; Williams, A.J.; Lai, F.A.; Zissimopoulos, S. Calsequestrin interacts directly with the cardiac ryanodine receptor luminal domain. J. Cell Sci. 2016, 129, 3983–3988.
- Zhang, L.; Kelley, J.; Schmeisser, G.; Kobayashi, Y.; Jones, L. Complex formation between Junctin, Triadin, Calsequestrin, and the ryanodine receptor: Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J. Biol. Chem. 1997, 272, 23389–23397.
- Sibbles, E.T.; Waddell, H.M.M.; Mereacre, V.; Jones, P.P.; Munro, M.L. The function and regulation of calsequestrin-2: Implications in calcium-mediated arrhythmias. Biophys. Rev. 2022, 14, 329–352.
- Knollmann, B.C.; Chopra, N.; Hlaing, T.; Akin, B.; Yang, T.; Ettensohn, K.; Knollmann, B.E.; Horton, K.D.; Weissman, N.J.; Holinstat, I.; et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J. Clin. Investig. 2006, 116, 2510–2520.
- Song, L.; Alcalai, R.; Arad, M.; Wolf, C.M.; Toka, O.; Conner, D.A.; Berul, C.I.; Eldar, M.; Seidman, C.E.; Seidman, J.G. Calsequestrin 2 (CASQ2) mutations increase expression of calreticulin and ryanodine receptors, causing catecholaminergic polymorphic ventricular tachycardia. J. Clin. Investig. 2007, 117, 1814–1823.
- Gray, B.; Bagnall, R.D.; Lam, L.; Ingles, J.; Turner, C.; Haan, E.; Davis, A.; Yang, P.C.; Clancy, C.E.; Sy, R.W.; et al. A novel heterozygous mutation in cardiac calsequestrin causes autosomal dominant catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2016, 13, 1652–1660.
- Ng, K.; Titus, E.W.; Lieve, K.V.; Roston, T.M.; Mazzanti, A.; Deiter, F.H.; Denjoy, I.; Ingles, J.; Till, J.; Robyns, T.; et al. An International Multicenter Evaluation of Inheritance Patterns, Arrhythmic Risks, and Underlying Mechanisms of CASQ2-Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2020, 142, 932–947.
- Titus, E.W.; Deiter, F.H.; Shi, C.; Wojciak, J.; Scheinman, M.; Jura, N.; Deo, R.C. The structure of a Calsequestrin filament reveals mechanisms of familial arrhythmia. Nat. Struct. Mol. Biol. 2020, 27, 1142–1151.
- Wleklinski, M.J.; Kannankeril, P.J.; Knollmann, B.C. Molecular and tissue mechanisms of catecholaminergic polymorphic ventricular tachycardia. J. Physiol. 2020, 598, 2817–2834.
More