Multiple sclerosis (MS) is an inflammatory demyelinating autoimmune disease of the central nervous system (CNS) that typically manifests with episodes of new/recurrent neurological symptoms (i.e., relapses) followed by either complete remission or residual disability (relapsing–remitting, RR, course).
Recent evidence of effectiveness of B cell-depleting monoclonal antibodies (mAbs) in MS prompted a partial revisitation of the pathogenetic paradigm of the disease, which has been, so far, considered a T cell-mediated autoimmune disorder. Although mechanisms underlying the efficacy of B cell-depleting mAbs in MS are not fully elucidated, they likely involve the impairment of pleiotropic B cell functions different from antibody secretion, such as their role as antigen-presenting cells. A potential impact of B cell-depleting mAbs on inflammation compartmentalized within the central nervous system was also suggested, but little is known about the mechanism underlying this latter phenomenon.
- multiple sclerosis
- B cell-depleting therapy
- monoclonal antibody
1. Introduction
2. Insights into MS Pathogenesis: Onset of Autoimmunity
MS was traditionally considered a T-cell-mediated autoimmune disorder, based on preclinical data from animal models of the disease (experimental autoimmune encephalomyelitis—EAE) and evidence for T-cell infiltration in inflammatory lesions and normal-appearing white matter of autoptic and biopsy CNS specimens from affected individuals, with an association between CD8+ T cells number and axonal damage [5][6][7][8][9][10][11][12]. Furthermore, the identification of expanded T-cell clones in the brain parenchyma, cerebrospinal fluid (CSF), and peripheral blood of MS patients detected using T-cell receptor (TCR) analyses reinforced the hypothesis that inflammatory infiltrates were constituted by pathogenetic expanded T-cell clones reactive to myelin antigens [13][14][15][16]. Circulating CD4+ T cells from MS patients were indeed demonstrated to recognise myelin basic protein (MBP), proteolipid protein (PLP), and myelin oligodendrocyte glycoprotein (MOG), even if the same phenomenon was also observed in healthy individuals; evidence regarding potential differences between these groups in frequency and avidity of cell interactions is conflicting [17][18]. A contribution of B cells to MS pathogenesis was also suggested by preclinical and clinical evidence, and their role was recently re-evaluated with the observation of a remarkable therapeutic effect of B-cell-depleting strategies [19]. Innate immunity cells, including CNS-resident microglia, contribute to MS pathogenesis, and neurodegenerative phenomena possibly, at least in part, independent of inflammation play a role in advanced disease [3]. Although MS aetiology is unknown, several environmental and genetic risk factors were identified [20][21][22]. Class II major histocompatibility complex (MHCII) represents the major genetic risk factor, accounting for 20–30% of individual genetic susceptibility [22][23]. MHCII genes encode membrane glycoproteins that are expressed by professional antigen-presenting cells (APC), such as dendritic cells, macrophages, and B cells [24]. The MHCII complex plays a key role in the development of both primary and secondary T CD4-mediated immune response as it presents in its context small peptide antigens processed by professional APC to MHC-restricted T cells [25]. In addition to MHC, several other genes were associated with the risk of developing MS on the basis of a complex genetic background, as confirmed by genome-wide association studies (GWAS) that uncovered more than 200 genetic susceptibility variants which could jointly account for ~48% of the estimated heritability for MS [26]. Enrichment for MS susceptibility loci was mostly related to genes involved in immune system function and regulation, and it was apparent in many different immune cell types and tissues, including microglia, highlighting, overall, the relevance of adaptive and innate immune cells in MS pathogenesis.2.1. Primary Autoimmune Response in the Peripheral Compartment
The mechanisms which trigger MS are still debated and two plausibly complementary pathogenetic models were proposed to explain the initiation of the autoimmune response, suggesting that the primum movens might take place either in the periphery (“outside-in” model) or within the CNS (“inside-out” model) [27]. The “outside-in” (or CNS-extrinsic/peripheral) model resembles the pathogenetic mechanism underlying EAE in which the disease is induced by external immunisation obtained through the inoculation of myelin-specific antigens in combination with an adjuvant [28]. According to this model, activation and expansion of CNS antigen-specific CD4+ T cells occur in the periphery and may be induced by an encounter with exogenous antigens sharing structural motifs with myelin antigens, hence capable of eliciting an autoimmune response based on molecular mimicry [29][30]. Several infective agents were suggested as potential exogenous triggers of the autoimmune reaction, with Epstein–Barr virus (EBV) the most plausible candidate [31][32]. On the other hand, the “inside-out” model suggests that the autoimmune reaction is triggered by an “internal event” occurring within the CNS and generating myelin debris. CNS-derived soluble antigens may then be drained via lymphatic pathways to peripheral lymphoid organs, where they might be presented to T and B cells eliciting the autoimmune reaction [33][34]. Different hypotheses suggest that the CNS-intrinsic event might be triggered by a CNS viral infection or by primary neurodegeneration [11].2.2. Secondary Autoimmune Response in the Central Nervous System
Once their priming has occurred, autoreactive T cells might be further expanded in peripheral lymphoid tissues after the encounter with their cognate antigen [22][35]. In the absence of neuroinflammation, T cells patrol the CNS crossing the BBB at the level of CNS post-capillary venules and reaching perivascular or subarachnoid spaces [36]. In such areas, putative cross-reactive T-cell clones may be further activated by CNS antigens (sharing structural motifs with their cognate antigen) which, after being drained from the parenchyma, are processed and presented by tissue-resident APCs. This mechanism promotes a secondary immune response within the CNS characterised by cell proliferation, recruitment of pro-inflammatory cells, and formation of perivascular cuffs around post-capillary and pial venules [37]. Upon activation, adaptive immune cells cross the glia limitans and infiltrate the CNS parenchyma where they produce pro-inflammatory cytokines and chemokines that determine a breakdown of the BBB. This causes further recruitment of adaptive and innate immune cells, which ultimately promote the formation of demyelinating lesions and tissue injury (Figure 1) [37][38]. Further damage to CNS tissue might derive from uncovering and release in the extracellular space of several autoantigens which, in turn, might enhance the autoimmune response in a mechanism defined as epitope spreading [39].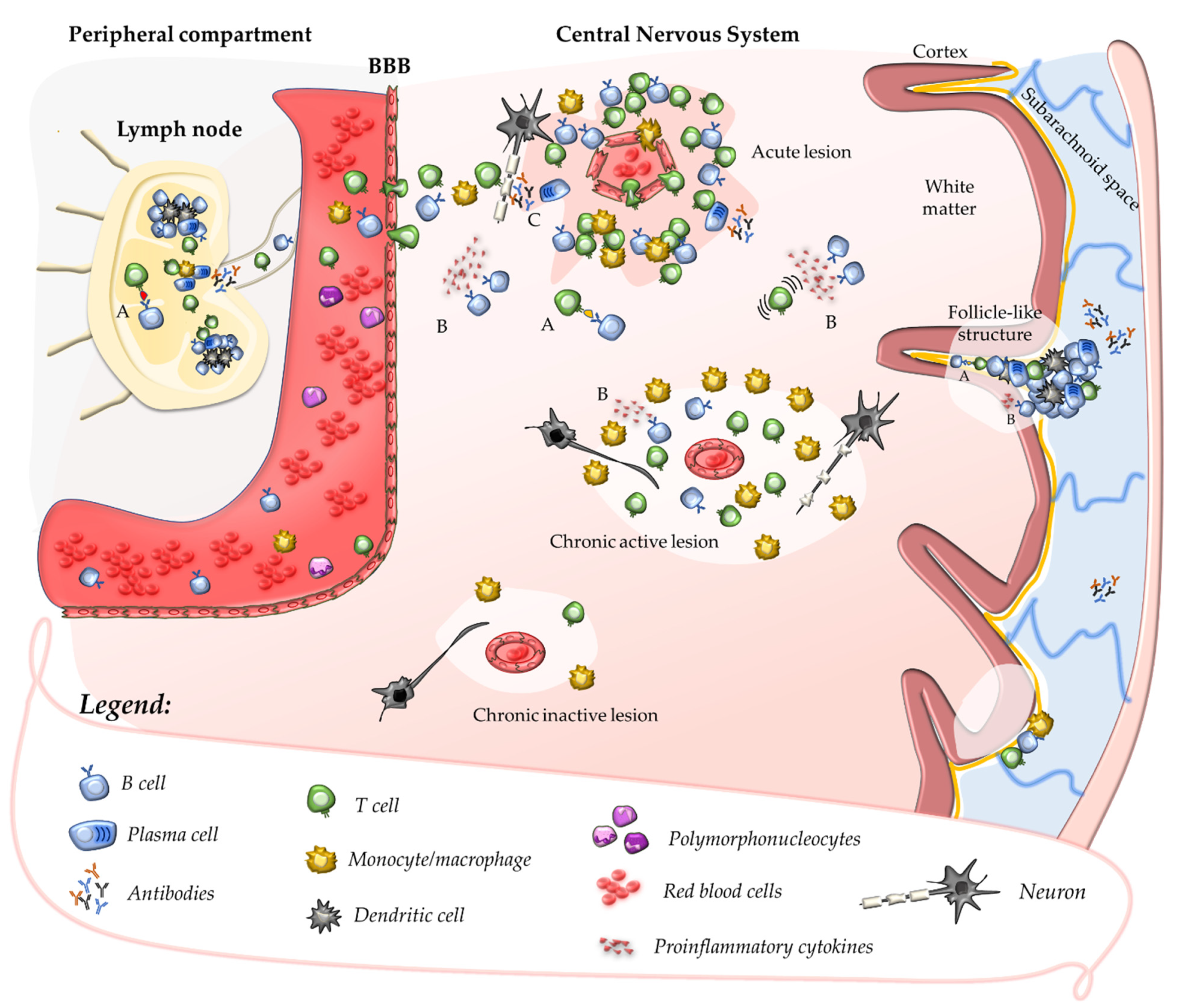
2.3. Contribution of B Cells and Humoral Response to Acute CNS Injury
The role of B cells and humoral immunity in the pathogenesis of MS was considered less prominent compared with that of T cells, possibly due to the lack of consistency in detecting antibodies specific to CNS self-antigens in brain lesions or CSF [43]. However, their contribution was recently reinforced by the observation of the remarkable effectiveness of monoclonal antibodies (mAbs) targeting this cell population [44]. Evidence for oligoclonal antibody production in the CSF dates to the 1940s [45], and an oligoclonal pattern of intrathecal production of immunoglobulins (OCBs) is detected in the CSF of the vast majority of MS patients; OCBs are plausibly produced by a restricted number of plasma cell clones recruited in the CNS during the secondary autoimmune response [46]. Experimental data derived from one of the animal models of MS more similar to humans—EAE induced by immunisation with myelin-antigens in marmosets—show that a secondary antigen-specific humoral immune response is elicited at a perivenular level by cellular immunity [47]. In the animal model, breakdown of the BBB in the surrounding capillaries is promoted by diffusive molecules secreted by immune cells aggregated as perivascular cuffs: in this setting, circulating antibodies specific to myelin antigens contribute to myelin damage [48]. However, differently from this model, circulating antibodies against CNS antigens are rarely detected in humans [49]; nevertheless, anatomopathological studies showing the deposition of immunoglobulins (mainly IgG) and complement C9neo antigen at sites of active myelin destruction in pattern II lesions suggest a possible contribution of the humoral response to tissue injury [50]. Pleiotropic roles of B cells independent of antibody secretion may be relevant to MS pathogenesis, such as antigen-presenting function or secretion of pro-inflammatory cytokines (Figure 1) [51]. Clonally expanded B-cell clones were detected in the meninges, parenchyma, and CSF of MS patients; they may indefinitely persist within perivascular and leptomeningeal inflammatory infiltrates where they organise in follicle-like tertiary lymphoid structures of aggregated plasma cells, B cells, T cells, and follicular dendritic cells [52][53][54]. Recent data suggest that B-cell accumulation within inflammatory infiltrates may be critical to the chronicisation of the intrathecal autoimmune response, acting as professional APCs for definite CNS autoantigens [55].References
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple sclerosis. Lancet 2018, 391, 1622–1636.
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286.
- Lassmann, H. Pathogenic Mechanisms Associated With Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2018, 9, 3116.
- Lassmann, H. Pathology and disease mechanisms in different stages of multiple sclerosis. J. Neurol. Sci. 2013, 333, 1–4.
- Babbe, H.; Roers, A.; Waisman, A.; Lassmann, H.; Goebels, N.; Hohlfeld, R.; Friese, M.; Schroder, R.; Deckert, M.; Schmidt, S.; et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J. Exp. Med. 2000, 192, 393–404.
- Junker, A.; Ivanidze, J.; Malotka, J.; Eiglmeier, I.; Lassmann, H.; Wekerle, H.; Meinl, E.; Hohlfeld, R.; Dornmair, K. Multiple sclerosis: T-cell receptor expression in distinct brain regions. Brain 2007, 130, 2789–2799.
- Skulina, C.; Schmidt, S.; Dornmair, K.; Babbe, H.; Roers, A.; Rajewsky, K.; Wekerle, H.; Hohlfeld, R.; Goebels, N. Multiple sclerosis: Brain-infiltrating CD8+ T cells persist as clonal expansions in the cerebrospinal fluid and blood. Proc. Natl. Acad. Sci. USA 2004, 101, 2428–2433.
- Markovic-Plese, S. Degenerate T-cell receptor recognition, autoreactive cells, and the autoimmune response in multiple sclerosis. Neuroscientist 2009, 15, 225–231.
- Montes, M.; Zhang, X.; Berthelot, L.; Laplaud, D.A.; Brouard, S.; Jin, J.; Rogan, S.; Armao, D.; Jewells, V.; Soulillou, J.P.; et al. Oligoclonal myelin-reactive T-cell infiltrates derived from multiple sclerosis lesions are enriched in Th17 cells. Clin. Immunol. 2009, 130, 133–144.
- Saxena, A.; Martin-Blondel, G.; Mars, L.T.; Liblau, R.S. Role of CD8 T cell subsets in the pathogenesis of multiple sclerosis. FEBS Lett. 2011, 585, 3758–3763.
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558.
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132, 1175–1189.
- Amoriello, R.; Chernigovskaya, M.; Greiff, V.; Carnasciali, A.; Massacesi, L.; Barilaro, A.; Repice, A.M.; Biagioli, T.; Aldinucci, A.; Muraro, P.A.; et al. TCR repertoire diversity in Multiple Sclerosis: High-dimensional bioinformatics analysis of sequences from brain, cerebrospinal fluid and peripheral blood. EBioMedicine 2021, 68, 103429.
- Amoriello, R.; Greiff, V.; Aldinucci, A.; Bonechi, E.; Carnasciali, A.; Peruzzi, B.; Repice, A.M.; Mariottini, A.; Saccardi, R.; Mazzanti, B.; et al. The TCR Repertoire Reconstitution in Multiple Sclerosis: Comparing One-Shot and Continuous Immunosuppressive Therapies. Front Immunol. 2020, 11, 559.
- Gestri, D.; Baldacci, L.; Taiuti, R.; Galli, E.; Maggi, E.; Piccinni, M.P.; Vergelli, M.; Massacesi, L. Oligoclonal T cell repertoire in cerebrospinal fluid of patients with inflammatory diseases of the nervous system. J. Neurol. Neurosurg. Psychiatry 2001, 70, 767–772.
- Martin, R.; Jaraquemada, D.; Flerlage, M.; Richert, J.; Whitaker, J.; Long, E.O.; McFarlin, D.E.; McFarland, H.F. Fine specificity and HLA restriction of myelin basic protein-specific cytotoxic T cell lines from multiple sclerosis patients and healthy individuals. J. Immunol. 1990, 145, 540–548.
- Bielekova, B.; Sung, M.H.; Kadom, N.; Simon, R.; McFarland, H.; Martin, R. Expansion and functional relevance of high-avidity myelin-specific CD4+ T cells in multiple sclerosis. J. Immunol. 2004, 172, 3893–3904.
- Hellings, N.; Baree, M.; Verhoeven, C.; D’Hooghe, M.B.; Medaer, R.; Bernard, C.C.; Raus, J.; Stinissen, P. T-cell reactivity to multiple myelin antigens in multiple sclerosis patients and healthy controls. J. Neurosci. Res. 2001, 63, 290–302.
- del Pilar Martin, M.; Cravens, P.D.; Winger, R.; Kieseier, B.C.; Cepok, S.; Eagar, T.N.; Zamvil, S.S.; Weber, M.S.; Frohman, E.M.; Kleinschmidt-DeMasters, B.K. Depletion of B lymphocytes from cerebral perivascular spaces by rituximab. Arch. Neurol. 2009, 66, 1016–1020.
- Sawcer, S.; Franklin, R.J.; Ban, M. Multiple sclerosis genetics. Lancet Neurol. 2014, 13, 700–709.
- Amato, M.P.; Derfuss, T.; Hemmer, B.; Liblau, R.; Montalban, X.; Soelberg Sorensen, P.; Miller, D.H.; Group, E.F.W. Environmental modifiable risk factors for multiple sclerosis: Report from the 2016 ECTRIMS focused workshop. Mult. Scler. 2017, 24, 590–603.
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768.
- Jersild, C.; Svejgaard, A.; Fog, T. HL-A antigens and multiple sclerosis. Lancet 1972, 1, 1240–1241.
- Ryan, S.O.; Cobb, B.A. Roles for major histocompatibility complex glycosylation in immune function. Semin. Immunopathol. 2012, 34, 425–441.
- Neefjes, J.; Jongsma, M.L.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836.
- International Multiple Sclerosis Genetics, Consortium; ANZgene; IIBDGC; WTCCC2. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365.
- Titus, H.E.; Chen, Y.; Podojil, J.R.; Robinson, A.P.; Balabanov, R.; Popko, B.; Miller, S.D. Pre-clinical and Clinical Implications of “Inside-Out” vs. “Outside-In” Paradigms in Multiple Sclerosis Etiopathogenesis. Front. Cell. Neurosci. 2020, 14, 599717.
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106.
- Harkiolaki, M.; Holmes, S.L.; Svendsen, P.; Gregersen, J.W.; Jensen, L.T.; McMahon, R.; Friese, M.A.; van Boxel, G.; Etzensperger, R.; Tzartos, J.S.; et al. T cell-mediated autoimmune disease due to low-affinity crossreactivity to common microbial peptides. Immunity 2009, 30, 348–357.
- Haring, J.S.; Pewe, L.L.; Perlman, S. Bystander CD8 T cell-mediated demyelination after viral infection of the central nervous system. J. Immunol. 2002, 169, 1550–1555.
- Sospedra, M.; Martin, R. Immunology of Multiple Sclerosis. Semin. Neurol. 2016, 36, 115–127.
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301.
- ’t Hart, B.A.; Luchicchi, A.; Schenk, G.J.; Stys, P.K.; Geurts, J.J.G. Mechanistic underpinning of an inside–out concept for autoimmunity in multiple sclerosis. Ann. Clin. Transl. Neurol. 2021, 8, 1709–1719.
- Laman, J.D.; Weller, R.O. Drainage of cells and soluble antigen from the CNS to regional lymph nodes. J. Neuroimmune Pharmacol. 2013, 8, 840–856.
- Mundt, S.; Greter, M.; Flugel, A.; Becher, B. The CNS Immune Landscape from the Viewpoint of a T Cell. Trends Neurosci. 2019, 42, 667–679.
- Mapunda, J.A.; Tibar, H.; Regragui, W.; Engelhardt, B. How Does the Immune System Enter the Brain? Front. Immunol. 2022, 13, 805657.
- Owens, T.; Bechmann, I.; Engelhardt, B. Perivascular spaces and the two steps to neuroinflammation. J. Neuropathol. Exp. Neurol. 2008, 67, 1113–1121.
- Kawakami, N.; Flügel, A. Knocking at the brain’s door: Intravital two-photon imaging of autoreactive T cell interactions with CNS structures. Semin. Immunopathol. 2010, 32, 275–287.
- Croxford, J.L.; Olson, J.K.; Miller, S.D. Epitope spreading and molecular mimicry as triggers of autoimmunity in the Theiler’s virus-induced demyelinating disease model of multiple sclerosis. Autoimmun. Rev. 2002, 1, 251–260.
- Scalfari, A.; Neuhaus, A.; Degenhardt, A.; Rice, G.P.; Muraro, P.A.; Daumer, M.; Ebers, G.C. The natural history of multiple sclerosis: A geographically based study 10: Relapses and long-term disability. Brain 2010, 133, 1914–1929.
- Filippi, M.; Rocca, M.A.; Ciccarelli, O.; De Stefano, N.; Evangelou, N.; Kappos, L.; Rovira, A.; Sastre-Garriga, J.; Tintorè, M.; Frederiksen, J.L. MRI criteria for the diagnosis of multiple sclerosis: MAGNIMS consensus guidelines. Lancet Neurol. 2016, 15, 292–303.
- Absinta, M.; Sati, P.; Schindler, M.; Leibovitch, E.C.; Ohayon, J.; Wu, T.; Meani, A.; Filippi, M.; Jacobson, S.; Cortese, I.C.; et al. Persistent 7-tesla phase rim predicts poor outcome in new multiple sclerosis patient lesions. J. Clin. Investig. 2016, 126, 2597–2609.
- Weber, M.S.; Hemmer, B.; Cepok, S. The role of antibodies in multiple sclerosis. Biochim. Biophy. Acta 2011, 1812, 239–245.
- Li, R.; Patterson, K.R.; Bar-Or, A. Reassessing B cell contributions in multiple sclerosis. Nat. Immunol. 2018, 19, 696–707.
- Kabat, E.A.; Moore, D.H.; Landow, H. An electrophoretic study of the protein components in cerebrospinal fluid and their relationship to the serum proteins. J. Clin. Investig. 1942, 21, 571–577.
- Correale, J.; de los Milagros Bassani Molinas, M. Oligoclonal bands and antibody responses in multiple sclerosis. J. Neurol. 2002, 249, 375–389.
- Gaitan, M.I.; Maggi, P.; Wohler, J.; Leibovitch, E.; Sati, P.; Calandri, I.L.; Merkle, H.; Massacesi, L.; Silva, A.C.; Jacobson, S.; et al. Perivenular brain lesions in a primate multiple sclerosis model at 7-tesla magnetic resonance imaging. Mult. Scler. 2014, 20, 64–71.
- Hart, B.A.; Massacesi, L. Clinical, pathological, and immunologic aspects of the multiple sclerosis model in common marmosets (Callithrix jacchus). J. Neuropathol. Exp. Neurol. 2009, 68, 341–355.
- Genain, C.P.; Cannella, B.; Hauser, S.L.; Raine, C.S. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat. Med. 1999, 5, 170–175.
- Lucchinetti, C.; Bruck, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000, 47, 707–717.
- Cencioni, M.T.; Mattoscio, M.; Magliozzi, R.; Bar-Or, A.; Muraro, P.A. B cells in multiple sclerosis-from targeted depletion to immune reconstitution therapies. Nat. Rev. Neurol. 2021, 17, 399–414.
- Lovato, L.; Willis, S.N.; Rodig, S.J.; Caron, T.; Almendinger, S.E.; Howell, O.W.; Reynolds, R.; O’Connor, K.C.; Hafler, D.A. Related B cell clones populate the meninges and parenchyma of patients with multiple sclerosis. Brain 2011, 134, 534–541.
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004, 14, 164–174.
- Howell, O.W.; Reeves, C.A.; Nicholas, R.; Carassiti, D.; Radotra, B.; Gentleman, S.M.; Serafini, B.; Aloisi, F.; Roncaroli, F.; Magliozzi, R.; et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011, 134, 2755–2771.
- Wang, J.; Jelcic, I.; Muhlenbruch, L.; Haunerdinger, V.; Toussaint, N.C.; Zhao, Y.; Cruciani, C.; Faigle, W.; Naghavian, R.; Foege, M.; et al. HLA-DR15 Molecules Jointly Shape an Autoreactive T Cell Repertoire in Multiple Sclerosis. Cell 2020, 183, 1264–1281.e20.