The heart needs metabolic flexibility, which allows the utilization of different substrates (FAs, carbohydrates, amino acid and ketones) to maintain the contractile function in response to energy stress. Within the heart, metabolic flexibility is governed by (1) substrate availability, and (2) a complex regulatory metabolic mechanism.
2. Metabolic Inflexibility as the Basis of Pathogenesis among AF Stressors
Metabolic inflexibility is common in pathological conditions. It has been reported that metabolic abnormalities in adults, including obesity, insulin resistance and/or T2DM, induce a state of impaired metabolic flexibility
[18][48]. Stull et al. showed that insulin resistance is a major contributor to metabolic flexibility in humans, and metabolic flexibility is negatively associated with aging
[19][58]. In addition, metabolic inflexibility is also evident in the failing heart
[20][8].
Therefore,
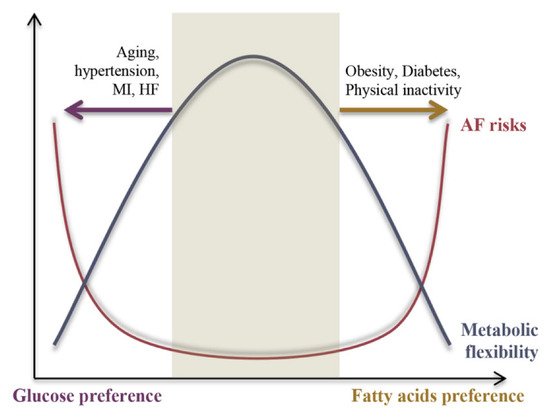
we propose a negative relationship between AF risk and metabolic flexibility
was proposed, as shown in
Figure 12. Under physiological conditions, the heart is highly metabolically flexible, which is associated with lower AF risk, whereas under pathological conditions metabolic flexibility is impaired and causes the energy substrate preference to switch either towards FAs or glucose, resulting in increased AF risk. The underlying mechanisms of the switch in energy substrate preference contributing to metabolic inflexibility will be discussed in the following section.
Figure 12.
A negative correlation between the AF risk and cardiac metabolic flexibility.
3. The Substrate-Metabolism Mechanism Underlying Metabolic Inflexibility
3.1. Substrate Metabolic Flexibility
Metabolic inflexibility has been implicated in decreased FAO capacity in FAs’ metabolism and insulin resistance and/or impaired insulin signaling in glucose metabolism. In the following,
we will discuss how the alterations in each substrate metabolism (glucose, FAs, amino acids and ketones) and their regulatory signaling affect metabolic flexibility
will be discussed.
3.1.1. Glucose Metabolic Inflexibility Underlying AF
There is increasing evidence suggesting that abnormal glucose metabolism is crucial for AF pathogenesis. Diabetes was shown to be correlated with a 34% greater risk of developing AF in a meta-analysis
[21][52], and to increase by 3% for each additional year of treatment
[22][86]. Epidemic studies have shown that hyperglycemia is also associated with an increased risk of AF
[22][23][86,87]. In addition, glucose fluctuations have been reported to play a key role in AF pathogenesis, as evidenced in diabetic rats
[24][88] and human subjects with diabetes
[25][89]. Chao et al. demonstrated an increase in the activation time of both atria and a decrease in bipolar voltage in patients with abnormal glucose metabolism, compared with those without
[26][90].
Insulin resistance, a key component of metabolic inflexibility, has been suggested as an independent risk factor for incident AF even before diabetes develops. However, some controversies regarding the role of insulin resistance in the epidemiology of AF still remain. Increasing numbers of studies have shown that metabolic syndrome, of which insulin resistance is a key component, is highly associated with AF risk. Lee reported that high values of insulin resistance assessed by homeostasis model (HOMA-IR), an insulin resistance index, were significantly associated with an increased risk of AF independent of other known risk factors in nondiabetic subjects
[27][91]. Conversely, several studies showed neutral or even opposite results. A cohort study proposed a negative relationship between fasting plasma insulin levels and AF risk
[28][92]. Several large cohort studies reported no independent association between insulin resistance and AF development
[29][30][93,94].
However, emerging animal studies have identified the positive relationship between IR and AF risk. Chan et al. showed an increase in AF susceptibility in insulin-resistant rats fed high-fat and high-fructose/cholesterol diets for 15 weeks
[31][95]. In addition, the loss of insulin signaling contributed to increased AF risk in Type 1 diabetic mice, and insulin treatment reduced AF susceptibility
[32][33][96,97].
Glucose metabolic flexibility relies on the configuration of metabolic pathways that manage glucose availability, uptake, glycolysis and glucose oxidation.
The pro-glycolysis effect in pathological conditions is always correlated to impaired insulin sensitivity. Insulin resistance is connected with abnormalities in switching between lipid and glucose utilization, thus acting as a key component of metabolic inflexibility. The potential mechanism underlying insulin-resistance-induced AF is associated with the decreased expression of Na
V1.5 and sodium current (I
Na)
[32][96], the abnormal up-regulation of calcium-homeostasis-related proteins (CaMKII)
[31][95], and the decreased expression and membrane translocation of glucose transporter type 4 (GLUT4) and GLUT8
[34][98] in an insulin-resistant state.
It was reported that glucose uptake and glycolysis are markedly increased in the failing heart, whereas glucose oxidation and mitochondrial function are decreased, indicating the uncoupling of glycolysis and glucose oxidation
[20][8]. This phenomenon is much like the “Warburg effect” commonly observed in rapidly growing tumor cells. Liu et al. reviewed and confirmed the existence of the Warburg effect in AF
[35][99]. The signaling pathway involved in the Warburg effect during AF includes the anti-Warburg-effect AMPK and pro-Warburg-effect pyruvate dehydrogenase kinase (PDK) and HIF-1α.
Unlike AMPK, the negative regulator of the Warburg effect, which is widely accepted to promote metabolic flexibility and decrease AF risk, pro-Warburg PDK and HIF-1α, is underestimated in their role in AF pathogenesis.
PDK is a key regulator of glycolysis–GO coupling by phosphorylating and inactivating the pyruvate dehydrogenase (PDH), leading to increased glycolysis and FAs’ metabolism. The heart needs to oxidize enough carbohydrate to meet energy needs, and previous reports showed that PDK overexpression can cause a loss of metabolic flexibility and exacerbate cardiomyopathy
[36][100]. In concert, the genetic inactivation of PDK4 improves hyperglycemia and insulin resistance
[37][101]. AF is associated with stimulated PDK expression. The specific inhibition of PDK4 via Dichloroacetic acid (DCA) can attenuate metabolic stress, myocardial fibrosis remodeling, and atrial intracardiac waveform activity in a paroxysmal canine model of AF
[38][102].
HIF-1α is a transcription factor driving the transcription of a variety of glycolysis-related genes, including PDK, GLUT1, hexokinase II (HKII), and lactate dehydrogenase A (LDHA), thus serving as a key regulator of glycolysis.
It has been reported that the ratio between glycolytic and oxidative enzyme activities is correlated negatively with insulin sensitivity
[39][103], indicating the pivotal role of glycolysis in metabolic flexibility. However, the role of the Warburg effect in AF pathogenesis has been under-estimated.
3.1.2. FAs Metabolic Inflexibility Underlying AF Pathogenesis
FAs are the preferred substrate of the heart, especially the atria. Excessive FA availability is highly relevant to AF. An increased circulating level of FAs is found in multiple metabolic AF etiologies, including obesity, T2DM
[40][104], HF
[41][105], and aging
[42][106]. In a prospective cohort study, plasma phospholipid 16:0, the most abundant saturated FA in diet and circulation, was positively associated with incident AF
[43][107]. In another observational study, high levels of circulating ceramides and sphingomyelins with palmitic acid (C16:0) appeared to increase the risk of incident AF
[44][108]. A recent metagenomic sequencing and metabolomics study demonstrated higher levels of plasma palmitic acid and oleic acid (C18:0) in AF patients relative to healthy individuals
[40][104].
FAs’ metabolic flexibility relies on the configuration of the metabolic pathways that manage FA availability, uptake, transportation and oxidation.
Accumulating studies have attested that disrupted FA metabolism induces atrial metabolic inflexibility in the complex pathophysiology of AF. Atrial FAs’ metabolic inflexibility was further explained as the permanent augment of CD36 to the sarcolemma that leads to chronic FA overloading, as occurs in a variety of AF etiologies, such as IR, obesity
[45][109], rapid atrial pacing
[46][110], and aging
[47][111]. Besides FA uptake, decreased FA transport and oxidation also contribute to FA overload. In concert, post-operation AF patients showed a repressed atrial expression of fatty acid binding protein 3 (FABP3), indicating the impairment of cytosol FA transportation
[48][112]. In the atria of permanent AF patients, the gene
[49][31] and protein
[50][113] levels of FAO-related enzymes were reduced. In addition,
theour results demonstrate that restoring FAO, targeting carnitine palmitoyltransferase-1B (CPT-1B) via L-carnitine (its endogenous cofactor), could attenuate obesity-induced AF
[51][81].
Physiologically, FAO capacity is coupled with FA uptake. However, under pathological conditions, the coupling between FAs uptake and FAO is disrupted, thereby inducing lipid accumulation and subsequent atrial lipo-toxicity
[52][114]. Lipo-toxicity is commonly associated with risk factors for AF, including IR, obesity, aging, and myocardial ischemia–reperfusion, caused by toxic lipid intermediates, including long-chain acyl-CoAs, lipid peroxides, ceramides, diacylglycerols and acyl-carnitines. Atrial lipo-toxicity elicits mitochondrial and endoplasmic reticulum dysfunctions, activates apoptotic cell death signaling, and interferes with insulin-stimulated glycogen uptake, which together engage mechanisms underlying alterations in the atrial anatomy (hypertrophy and fibrosis
[52][114]) and electrophysiology (connexin 43 lateralization, conduction propagation impairment
[53][115]), culminating in AF.
The FAs metabolic disorders, characterized by the uncoupling of FAs uptake and utilization, are divided into two classes: lipid accumulation and resultant lipo-toxicity, or over-activated mitochondrial β-oxidation. Of note, these two pathological conditions are both associated with impaired glucose metabolism and/or insulin signaling, as postulated by the “Randle cycle”. Lipid accumulation and lipo-toxicity aggravates insulin sensitivity and inhibits glycolysis via lipid intermediates such as ceramides and diacylglycerols
[54][116]. Over-activated mitochondrial β-oxidation inhibits glucose oxidation via increased acetyl-CoA and NADH/NAD+ ratio, and inhibited glycolysis, targeting PK and PFK, via increasing citrate
[55][117].
The role of diverse FAs metabolism-related enzymes and regulators in metabolic inflexibility-induced AF are still under debate, especially these involved in triglyceride turnover (diacylglycerol transferase/DAGT, acetyl-CoA carboxylase/ACC, and adipose triglyceride lipase/ATGL), FAO (malonyl-CoA decarboxylase/MCD and carnitine acetyltransferase/CrAT) and FAs delivery (FABP). In addition, little is known about the actual degree to which lipid handling is disrupted under physiological and pathological stimulations in the atrium with AF and AF risk factors.
3.1.3. Amino Acids’ Metabolic Flexibility
Branched-chain amino acids (BCAA), including leucine, isoleucine and valine, are the only amino acids that can be utilized as a source of energy generation in the TCA. BCAA have been acknowledged as bio-energetic fuel for protein synthesis and cell growth, and as bio-active molecules regulating nutrient-sensitive pathways involved in multiple metabolic processes.
A dysregulated BCAA metabolism confers a high degree of risk for cardiovascular diseases
[56][118] and was recently revealed to have clinical and experimental relevance to AF. The circulating BCAA level is increased in metabolic AF stressors including IR, obesity, and HF individuals
[57][119]. In an ongoing study (unpublished),
it w
ase found that an 8-week BCAA supplement (0.75%) can significantly enhance AF invincibility and increase left atrial volume in mice. Likewise, atrial BCAA catabolic deficiency can promote angiotensin II (Ang II)-induced AF and atrial fibrosis
[58][120], and underlie myocardial fibrosis and hypertrophy via the PI3K-AKT-mTOR pathway
[59][121]. These results can be explained by the elevated accumulation of toxic BCAA or their derivatives (such as branched-chain α-keto acids), which induces mitochondrial oxidative stress by enhancing superoxide production, inhibiting mitochondrial complex I, and reducing superoxide dismutase (SOD) activity
[60][122].
Although the specific role of BCAA catabolism in cardiac substrate metabolism is underexplored, recent studies on extra-cardiac tissues (pancreas, adipose, and skeletal muscle) have indicated that the quantity and proportion of circulating BCAA can modulate glucose, lipid, and protein handling
[57][119]. It has been reported that elevated circulating levels of BCAA are positively associated with metabolic disorder and insulin resistance
[61][123]. Mechanistically, (1) the BCAA-induced activation of mammalian target of rapamycin complex 1 (mTORC1) and the resultant negative feedback regulation of insulin signaling, and (2) the accumulation of toxic BCAA metabolites and the resultant mitochondrial dysfunction could be two plausible explanations linking BCAA and insulin resistance
[62][124].
Restoring BCAA metabolic flexibility is an under-appreciated anti-AF metabolic strategy, and the underlying mechanism and the independent impacts of different AA components await further validation.
3.1.4. Ketones and Metabolic Flexibility
Ketones, namely β-hydroxybutyrate (β-OHB), acetoacetate (AcAc) and acetone, are initially synthesized in liver mitochondria through “ketogenesis” and can circulate to extrahepatic tissues for terminal oxidation. Serum β-OHB, the predominate (70%) circulating form of ketones, can be transported into cardiomyocytes via mono-carboxylate transporters (MCT1 and MCT2) or simple diffusion, then converted to acetyl-CoA via β-OHB dehydrogenase (BDH1) and succinyl-CoA:3-oxoacid-CoA-transferase (SCOT) in the mitochondrial matrix, and finally enter into the TCA cycle to generate ATP
[63][125].
The heart utilizes ketones in proportion to their delivery determined by circulating content, thus generally make a minor contribution to cardiac energy supply under basal conditions (<3%)
[64][126]. Hence, cardiac ketone metabolism is augmented in parallel to stimulated hepatic ketogenesis, as occurs in the nutrient deprivation or diminished carbohydrate availability that accompanies encompassing starvation/fasting, exercise, and ketogenic diets
[65][127]. Interestingly, most of the AF risk factors, such as diabetes/IR
[65][127], congestive HF
[66][67][128,129], and dilated and hypertrophic cardiomyopathies
[68][130], can stimulate hepatic ketogenesis, which is presumably associated with cardiac dysfunction, hemodynamic abnormalities and increased neuro-hormonal stress-related lipolysis. In a metabolomics profiling analysis of atria in AF patients, the concentrations of β-OHB, tyrosine, leucine (ketogenic AA) and fumarate (a metabolic intermediate in the TCA cycle) were reported to be elevated, indicating that ketone metabolism might be up-regulated in AF
[69][131].
Ketones are an alternative energy source in the energetically compromised heart; and are thus capable of promoting metabolic flexibility. Supportively, ketones can improve energy efficiency in the failing heart
[68][130], and decrease the infarct size and myocardial cell death following ischemic injury
[70][132]. These findings are further explained by ketones’ oxygen-efficient nature, which improves cardiac cell excitation–contraction coupling during hypoxia, as evidenced by its higher P/O ratio (2.50) relative to FAs (2.33), and the ability to inhibit FAO and accelerate the mitochondrial energy transduction of GO in working rat hearts
[71][133]. However, the role of ketones in regulating glucose metabolism and insulin sensitivity is equivocal, and was reviewed in
[72][134]. Generally, a short-term ketone diet improved glucose metabolism and insulin sensitivity, whereas a long-term ketone diet showed neutral or negative results
[73][74][135,136].
More importantly, the metabolic modification targeting ketone metabolism is complicated in AF patients, since a long-term up-regulated ketone metabolism can impose potential arrhythmogenic risks. Supportively, β-OHB can prolong the action potential by blocking the I
to in murine ventricular cardiomyocytes, and suppress sympathetic nervous system (SNS) activity to reduce the heart rate and cardiac energy expenditure by antagonization via G protein-coupled receptor 41 (GPR41)
[75][137]. In addition, a recent rodent study demonstrated that long-term exogenous β-OHB administration (16 weeks) can induce cardiac fibrosis and cellar apoptosis by inhibiting mitochondrial biogenesis
[76][138]. Ketones and intermediate metabolites (such as acetyl-CoAs) are also understudied metabolic signals that regulate the provision of fuel and mitochondrial energetics
[77][78][139,140]; thus, the alteration of ketone-derived signal components possibly influences the metabolic status to confer a metabolic risk of AF.
Given recent evidence, up-regulated ketone metabolism might be an emergency anti-AF strategy by remedying metabolic flexibility, but is not applicable for long-term AF management. A deeper understanding of ketone metabolism in AF with different etiologies is urgent in the field of AF.
3.2. Metabolism Regulatory Signaling and Metabolic Flexibility
The inactivation of AMPK is highly relevant to AF, as evidenced by the increase in AF vulnerability in various pre-clinical AF models and in genetically modified strains (Cardiac LKB1 KO mice)
[79][142]. The pharmacological activation of AMPK and its downstream regulator, PGC-1α, can retard/reverse the pathological processes underlying AF in human
[80][81][143,144] and pre-clinical AF models induced by rapid atrial pacing
[82][83][84][145,146,147] and obesity
[51][81], in addition to genetic modified rodents
[85][148].
Besides AMPK, a number of signaling pathways, including PGC-1α, sirtuins, and FOXO, HIF-1α, PPAR, and Akt, are also implicated in the regulation of metabolic flexibility. Their roles in metabolic flexibility and the pharmaceutical approaches to improving metabolic flexibility are beyond the scope of this
re
ntryview; please refer to
[2][86][7,149].
3.3. The Substrate-Metabolism Mechanism Underlying Metabolic Inflexibility and AF Pathogenesis
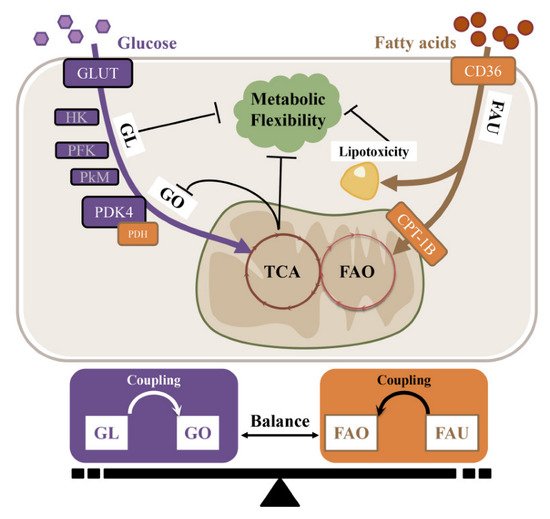
Under different forms of pathogenesis, the energy substrate preference shifts either towards (e.g., obesity and diabetes) or away from (e.g., aging, heart failure, and hypertension) FAO in the atria. As previously mentioned, the atria need metabolic flexibility to enable them to switch from fat to carbohydrate oxidation in response to AF and AF-induced ischemia. The overall (glucose–FAs utilization) and internal balance (glycolysis–glucose oxidation or FA uptake–FAO) of energy substrate metabolism is crucial for normal heart function (
Figure 23).
Figure 23. The substrate-metabolism mechanism underlying metabolic inflexibility. GL, Glycolysis; GO, Glucose oxidation; FAO, Fatty acid oxidation; FAU, Fatty acid uptake.
The uncoupling of glycolysis and oxidative phosphorylation is called “the Warburg effect”, and is mainly mediated via the up-regulation of PDK4 and the resultant inactivation of PDH activity. In hypoxic conditions such as aging, hypertension and HF, the metabolic balance shifts from oxidative phosphorylation to glycolysis to increase the efficiency of ATP produced in relation to oxygen consumed. It has been reported that the glycolysis rate is negatively associated with mitochondrial function and insulin sensitivity; thus, this metabolic switch to aerobic glycolysis leads to metabolic inflexibility. However, the relevant mechanism of glycolysis in insulin resistance and metabolic inflexibility warrants further investigation.
4. Anti-AF Strategies Targeting Metabolic Inflexibility
An AF strategy targeting metabolic inflexibility must consider the unique metabolic profile of certain AF etiologies, thus the recommendation of prevention and treatment for AF patients must be prudent and personal. To further elaborate the individualization of AF management, AF populations are divided into two categories based on their metabolic characteristics: (1) energy-rich pro-FAO condition (e.g., obesity and diabetes), and (2) energy-demanding pro-GL condition (e.g., aging, hypertension, myocardial ischemia, and HF).
When atrial cardiomyocytes are in an energy-rich pro-FAO condition such as obesity or T2DM, the atrium is characterized by over-whelming FAs intake and insufficient FAO in the atrium. The uncoupling of FAs uptake and FAO leads to lipid accumulation and lipo-toxicity, thereby inducing insulin resistance and metabolic inflexibility via toxic lipid intermediates.
When atrial cardiomyocytes are in an energy-starved/demanding pro-GL state such as aging, hypertension and HF, mitochondrial dysfunction occurs, and the TCA intermediate citrate can (1) inhibit the PDH activity and GO rate, and (2) block FAO by inhibiting CPT-1B through its derivation malonyl-CoA
[87][151]. Thus, the atrium usually enhances the metabolism of glucose or other alternative substrates (KBs) at the expense of diminished FAO to compensate for energy deficiency. Previous animal and human studies have shown that the altered preference for atrial fuel, namely metabolic remodeling, is responsible for inducing reversible electrical reconfiguration and the irreversible structural reconfiguration that predispose patients to AF occurrence and maintenance
[88][152]. Therefore, when treating this type of AF, moderate exercise, a heart-healthy diet, and pharmaceutical approaches to improve metabolic flexibility are of higher clinical relevance. For more details on the pharmaceutical approaches to improving metabolic flexibility, please refer to
[2][86][7,149].