Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Thulani Sibanda and Version 2 by Conner Chen.
Listeria monocytogenes is a foodborne pathogen that is the causative agent of the human disease, listeriosis. It is primarily a ubiquitous environmental saprophyte found in many environmental niches such as water, soil, and vegetation. Contaminated, often ready-to-eat (RTE) foods are the main transmission vehicles for human L. monocytogenes infections.
- Listeria monocytogenes
- virulence
- invasion
1. L. monocytogenes Virulence Factors
L. monocytogenes expresses several surface and soluble proteins that mediate the adhesion to target cells, internalization, intracellular multiplication and dissemination to other host cells [1][6]. The virulence factors are encoded either as separate loci across the bacterial genome or as clusters on pathogenicity islands [2][27]. A core of virulence genes (prfA, hly, actA, plcA, mpl, and plcB) encoded on the Listeria pathogenicity island 1 (LIPI-1) is conserved in the genomes of all L. monocytogenes strains [2][27]. Additionally, many other virulence factors encoded in separate loci, such as the internalin A/Internalin B (inlAB) operon, are also part of the virulence arsenal conserved in all L. monocytogenes strains [3][28]. The characteristics and roles of some of these proteins in the pathogenesis of L. monocytogenes are discussed in this section.
Listeria adhesion protein (LAP). LAP is a 104-kDa cell wall protein ubiquitously found in all Listeria species [4][29]. It was first described by Pandiripally et al. [5][30] as protein p104 which was subsequently found to be an alcohol acetaldehyde dehydrogenase [6][31]. As an essential enzyme, LAP is produced primarily as a cytosolic protein in all Listeria species. However, in pathogenic species, the protein is translocated to the cell surface through the SecA2 secretory system to facilitate the adhesion of pathogenic Listeria species to intestinal cells [7][8][32,33]. The epithelial receptor for LAP is a constitutively expressed mitochondrial protein, heat shock protein 60 (Hsp60) [4][29]. In addition to acting as an adhesin, LAP has also been implicated in the translocation of the pathogen across the intestinal epithelium [9][34].
Fibronectin binding protein (FbpA). Fibronectin binding proteins (Fbp) are cell wall-anchored proteins that are widely distributed in Gram-positive bacteria [10][35]. Fbps recognize and bind to fibronectin (a component of the human extracellular matrix that plays a role in inter-cellular interaction and adhesion) [11][36]. The interaction between bacterial Fbps and fibronectin molecules forms a three-component bridge (involving integrins), which facilitates the adhesion between the bacterial and the host cells [10][35]. The Fbp of L. monocytogenes (FbpA) was characterized by Dramsi et al. [12][37]. It is a 570-amino-acid polypeptide that shares a high homology to streptococcal Fbps (PavA of Streptococcus. pneumoniae, Fbp54 of S. pyogenes and FbpA of S. gordonii) [12][37]. However, unlike streptococcal Fbps, the L. monocytogenes FbpA is exposed on the surface of the bacterial cell without the signal peptide [12][37].
Internalin A (InlA). InlA is one of the principal virulence factors of L. monocytogenes that was first described by Gaillard et al. [13][38]. It is an 80 kDa protein that is anchored onto the cell wall peptidoglycan through a C-terminal LPXTG motif [14][39]. InlA mediates the adhesion and internalization of the pathogen into enterocytes in the first step of invasion of the intestinal barrier [15][22]. An N-terminal leucine-rich repeat (LRR) domain acts as the recognition and binding site to the EC1 domain of the extracellular portion of E-cadherin [16][17][40,41].
Internalin B (InlB). InlB is another adhesion protein that plays a major role in L. monocytogenes binding to enterocytes and the subsequent invasion of the intestinal barrier [14][39]. Unlike InlA, InlB is anchored onto the cell wall through glycine and tryptophan (GW) modules that non-covalently interact with cell wall teichoic acids [18][42]. The LRR domain acts as the recognition and binding site to Met (a host receptor tyrosine kinase) [15][22]. L. monocytogenes also produces many other LRR proteins classified under the internalin family [19][43]. However, InlA and InlB have been identified as the principal adhesion proteins that mediate pathogen binding and invasion [20][44].
Listeriolysin O (LLO). LLO is a 56 kDa pore-forming cytotoxin encoded by the hly gene [21][22][45,46]. It belongs to the family of cholesterol-dependent cytolysins (CDCs) [22][46]. It was one of the first L. monocytogenes virulence factors identified, based on the ability of virulent strains to cause hemolysis on blood agar [23][47]. Subsequent experiments identified the hemolysin as a sulfhydryl-activated toxin responsible for the intracellular growth of L. monocytogenes in human enterocyte-like Caco-2 cells [24][25][48,49]. The role of LLO is the lysis of the internalization vacuole, resulting in the release of the pathogen into the cytosol of host cells [26][50].
Phospholipases. Two types of phospholipases are required for L. monocytogenes. Phosphatidylinositol-specific phospholipase C (PI-PLC) is encoded by the plcA gene while phosphatidylcholine phospholipase C (PC-PLC) is encoded by the plcB gene [27][28][51,52]. PI-PLC plays a complementary role together with LLO in the lysis of the primary and secondary vacuole following pathogen internalization [20][44]. It catalyzes the cleavage of the membrane phosphatidylinositol into inositol phosphate and diacylglycerol [29][53]. PC-PLC is a broad-range phospholipase which is particularly required for the lysis of the double-membrane secondary vacuole and the primary vacuole in conditions of LLO deficiency [30][54]. PC-PLC is synthesized as a 33-kDa precursor that requires cleavage to produce the active 29-kDa enzyme [31][55]. A zinc-dependent metalloprotease (Mpl) encoded by the mpl gene is required for the maturation of PC-PLC [31][55].
Actin-polymerizing protein ActA. ActA is a surface protein encoded by the actA gene [32][56]. It mediates bacterial motility inside infected host cells through actin polymerization [32][56]. The protein is anchored on the bacterial cell membrane through its hydrophobic C-terminal domain while the functional N-terminal domain is exposed to the host cell cytoplasm [32][56]. Within the bacterial cell surface, ActA exhibits an asymmetrical distribution, being more concentrated at one polar end of the cell. The asymmetrical distribution is responsible for the directionality of L. monocytogenes motility [33][34][57,58]. To facilitate intracellular motility, ActA mediates actin nucleation and filament formation through the recruitment of host vasodilator-stimulated phosphoprotein (VASP) and actin-related proteins-2 and 3 (Arp2/3) complex [35][36][59,60].
2. Gastrointestinal Tract Colonization and Invasion of Host Cells
Due to its severity and high fatality rates, much of the focus on the pathogenesis of listeriosis is placed on invasive infections. However, evidence shows that non-invasive listerial febrile gastroenteritis outbreaks are very common [37][38][39][40][61,62,63,64]. Non-invasive L. monocytogenes infections are typically characterized by enteric symptoms such as vomiting, non-bloody diarrhea, nausea and fever that occur within a short period (24 h) following the ingestion of contaminated foods [38][41][62,65]. The mechanisms underlying the pathogenesis of non-invasive L. monocytogenes infections remain unclear [41][65]. Recently, a few studies have attempted to elucidate the mechanisms of L. monocytogenes gastrointestinal tract colonization [41][42][26,65]. Based on in vitro and mice models, the actin polymerization protein ActA—which mediates the cell-to-cell spread of the pathogen in invasive listeriosis—has also been implicated in intestinal colonization [42][26]. Using actA gene mutants in orally infected mice, Travier et al. [42][26] found that ActA can mediate L. monocytogenes aggregation both in vitro and in the gut lumen. The postulated mechanism of the ActA-mediated aggregation is based on direct ActA–ActA interactions through the C-terminal regions (which are not involved in polymerization) [42][26]. In the same study, the researchers found that ActA-dependent aggregation was also responsible for an increased ability to persist within the cecum and colon lumen of mice. Additionally, Halbedel et al. [41][65] observed a genetic correlation between the L. monocytogenes disease outcome (invasive or non-invasive) and the presence or absence of a functional chitinase gene (chiB) in which gastroenteritis outbreak isolates possessed a premature stop codon in the chiB gene. However, the restoration of chitinase production in a non-invasive isolate could not generate the invasiveness characteristic [41][65]. The first step in the pathogenesis of invasive listeriosis is the ability of the pathogen to cross the intestinal epithelial barrier. Although the complete mechanisms are still not fully understood, three well-elucidated pathways have thus far been used to explain the process [15][22]. These three pathways are the InlA-mediated transcytosis, the LAP-mediated translocation, and the microfold (M-cell)-mediated transcytosis [15][22]. InlA-mediated transcytosis. The InlA- mediated pathway is the primary route by which L. monocytogenes invades intestinal cells. InlA is a cell wall-anchored protein that mediates the uptake of L. monocytogenes into non-phagocytic cells through receptor-mediated endocytosis [43][66]. InlA promotes pathogen adhesion and the invasion of the intestinal epithelium through an interaction with its receptor, E-cadherin (a component of adherens junctions) [20][44]. Adherens junctions, tight junctions, and desmosomes are part of the apical junctional complex that provides a paracellular seal between adjacent epithelial cells [15][22]. The InlA interaction with receptors occurs at sites where E-cadherin is transiently exposed to the intestinal lumen [44][45][67,68]. The transient exposure of E-cadherin occurs during cell extrusion and junction remodeling [45][68]. Furthermore, changes in the shape of goblet cells can also result in the exposure of the E-cadherin component of the cell junctions [44][67]. Through interaction with the receptor, bacterial cells are taken into the enterocytes by endocytosis and are subsequently then released into the lamina propria by exocytosis [15][22]. The binding of InlA induces the recruitment of other junctional proteins, α-catenin and β-catenin, as well as actin and p120 catenin, which facilitate E-cadherin clustering at the site of bacterial entry [46][69]. Subsequently, a post-translational modification of E-cadherin (phosphorylation by the tyrosine kinase, Src and ubiquitination by the ubiquitin-ligase Hakai) induces endocytosis through caveolin or clathrin [15][46][22,69]. Ultimately, the InlA/E-cadherin-mediated endocytosis involves components of the host cytoskeleton that facilitate the formation of localized host cell membrane protrusions that force the formation of endocytic vesicles around the adherent bacteria cell [20][44]. It is now known that host cytoskeletal proteins involved in actin nucleation such as the Arp2/3 complex and VASP are activated in response to InlA binding to its receptors [14][47][39,70]. Unlike InlA, InlB does not play a major role in the invasion of intestinal cells [14][39]. However, together with InlA, it plays a role in the invasion of other tissues such as the liver, spleen, CNS and placenta [48][23]. The InlB receptor is the ubiquitous tyrosine kinase Met whose normal ligand is Hepatocyte Growth Factor (HGF) [20][44]. The binding of InlB to Met results in the autophosphorylation of the cytoplasmic tail of the Met proteins, initiating a reaction cascade that culminates in the localized polymerization of actin and internalization of bacterial cells in the same way as InlA [43][66]. LAP-mediated translocation. For a long time, the InlA-mediated pathway was established as the main route of L. monocytogenes traversal of the intestinal epithelium [44][45][46][67,68,69]. However, subsequent evidence that strains possessing non-functional InlA could cause infections in orally dosed mice and guinea pigs [49][50][71,72] showed that the pathogen can use alternative mechanisms to achieve intestinal invasion [9][34]. The surface protein, LAP, which was initially identified as an adhesin that facilitates the binding of L. monocytogenes to enterocytes, also contributes to the translocation of the pathogen across the intestinal epithelium [9][34]. The pathway of LAP-mediated invasion was elucidated by Drolia et al. [9][34] using a Caco-2 cell line and a mouse model. The researchers showed that LAP induces the intestinal epithelial barrier dysfunction as a mechanism of promoting bacterial translocation. The binding of LAP to its luminal receptor protein Hsp60 activates myosin light-chain kinase (MLCK) that mediates the opening of the intestinal barrier through the redistribution of junctional proteins, claudin-1, occludin, and E-cadherin [9][34]. These reactions cause the opening of tight junctions between neighboring enterocytes allowing L. monocytogenes translocation [9][15][22,34]. Furthermore, the LAP-mediated translocation is thought to be an important precursor event for the InlA-dependent invasion, as it potentially provides pathogen access to E-cadherin in exposed adherens junctions [9][34]. M-cell mediated transcytosis. The microfold (M) cells are specialized epithelial cells that survey the intestinal mucosa for any antigens as part of the mucosal immune response. They readily take up antigens from the intestinal mucosa and transcytose them across the intestinal epithelium to the lymphoid tissues of the Peyer’s patches [51][73]. This process also serves as a passive route for the transcytosis of pathogens into the basolateral side of the follicle-associated epithelium [52][74]. While the role of M-cells in the transcytosis of L. monocytogenes has been well established, the mechanism of the pathogen interaction with such cells is not fully understood [52][74]. Evidence from in vitro and orally infected mice models has shown that in the absence of InlA, L. monocytogenes rapidly accumulate in the Peyer’s patches [53][54][75,76]. The prevailing paradigm on the M-cell mediated pathway is that transcytosis occurs across the M cells through a vacuole [15][48][22,23]. However, Rey et al. [52][74] established that in addition to the rapid vacuolar transcytosis, L. monocytogenes also escapes to the cytosol of the M-cells by vacuolar rupture. Once in the M-cell cytosol, the pathogen can initiate a direct ActA-based M-cell-to-enterocyte spread [52][74].3. Intracellular Survival and Dissemination
The ability to cross the intestinal barrier provides the main gate of L. monocytogenes entry into the bloodstream. Due to its predilection for the CNS and the placenta in pregnant women, neurolisteriosis, maternofetal infection and septicemia are the main clinical manifestations of invasive listeriosis [55][77]. The high tropism of L. monocytogenes for these tissues is unclear. The possible explanation has been attributed to the presence of E-cadherin and Met, the two receptor proteins for InlA and InlB, respectively [56][7]. Because of the presence of Met in the human umbilical vein endothelial cells (HUVEC), L. monocytogenes can invade the human placenta through an InlB-dependent mechanism [57][78]. In the CNS, both receptors are expressed at the surface of choroid plexus epithelial cells and Met is additionally expressed at the brain endothelial cells of the blood-cerebrospinal fluid (CSF) and blood–brain barriers. Hence, the invasion of the CNS is facilitated by both InlA and InlB mechanisms [56][7]. Once internalized into the target cells in a primary vacuole, the next step in the infection cycle is the escape from the primary vacuole into the cell cytosol [58][79] (Figure 13). This vacuolar escape is mediated by the production of LLO [58][59][8,79]. This pore-forming cholesterol-dependent cytotoxin causes the rupture of the vacuole and release of the bacterial cells into the host cell cytosol [60][80]. In addition to LLO, L. monocytogenes also employs phospholipases, such as PI-PLC, that significantly enhance the lysis of the primary vacuole [28][52]. Following a period of intracellular replication inside infected cells, the production of ActA results in the formation of actin comet tails which facilitate bacterial motility inside the cells as well as the spread to uninfected cells through membrane protrusions [61][9]. The double membrane of the resulting secondary vacuole is degraded by LLO in collaboration with PC-PLC [61][9].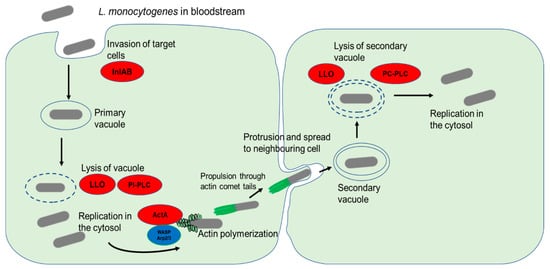
Figure 13. L. monocytogenes invasion of target cells and cell-to-cell spread. The bacterial surface internalins InlA and InlB interaction with their respective cell surface receptors result in the internalization of bacterial cells. The primary endocytic vacuole is then lysed through the activity of LLO and PI-PLC. Following a period of replication in the cytosol, the release of ActA stimulates actin polymerization by recruiting host nucleation proteins VASP and Arp2/3 complex. The formation of comet tails propels the bacterial cells and enables them to spread to neighboring cells through membrane protrusions. Lysis of the double membrane of the secondary vacuole by the action of LLO and PC-PLC causes the release of bacterial cells into the cytosol.