Perivascular adipose tissue (PVAT) is a special type of ectopic fat depot that adheres to most vasculatures. PVAT has been shown to exert anticontractile effects on the blood vessels and confers protective effects against metabolic and cardiovascular diseases. PVAT plays a critical role in vascular homeostasis via secreting adipokine, hormones, and growth factors. Endothelial nitric oxide synthase (eNOS; also known as NOS3 or NOSIII) is well-known for its role in the generation of vasoprotective nitric oxide (NO). eNOS is primarily expressed, but not exclusively, in endothelial cells, while recent studies have identified its expression in both adipocytes and endothelial cells of PVAT. PVAT eNOS is an important player in the protective role of PVAT.
- vascular function
- obesity
- nitric oxide
- adiponectin
- SIRT1
1. Introduction
2. What Is the Function of PVAT?
Since the first attention to the paracrine effects of PVAT on blood vessels [2], growing studies, from experimental animal models to clinical samples, have indicated that the cross-talk between PVAT and its connecting vessel plays a critical role in the physiological homeostasis and pathological changes of the cardiovascular system. The paracrine crosstalk between PVAT and its connecting vessel can actively regulate vascular inflammation and remodeling [23][7], while PVAT can also act as an endocrine organ to modulate multiple biological processes by releasing adipokines [25][8]. In 2002, using the physiological buffer in which PVAT from a healthy rat was incubated, Lohn et al. observed a direct relaxation in precontracted, isolated, PVAT-removed rat thoracic aorta [26][9]. They concluded the presence of transferable soluble substances from the PVAT that were released to the buffer and caused relaxation. It is currently known that PVAT is capable to synthesize and secrete substances via endocrine and paracrine mechanisms, including adipokines, growth factors, ROS, NO, and H2S [1]. The previous literature already explored the function of PVAT in detail [1,25,27,28,29,30][1][8][10][11][12][13]. Here, wresearchers briefly summarized the area of PVAT-derived adipokine production and vascular function regulation, and some novel findings of exosomes/extracellular vesicles (Figure 1).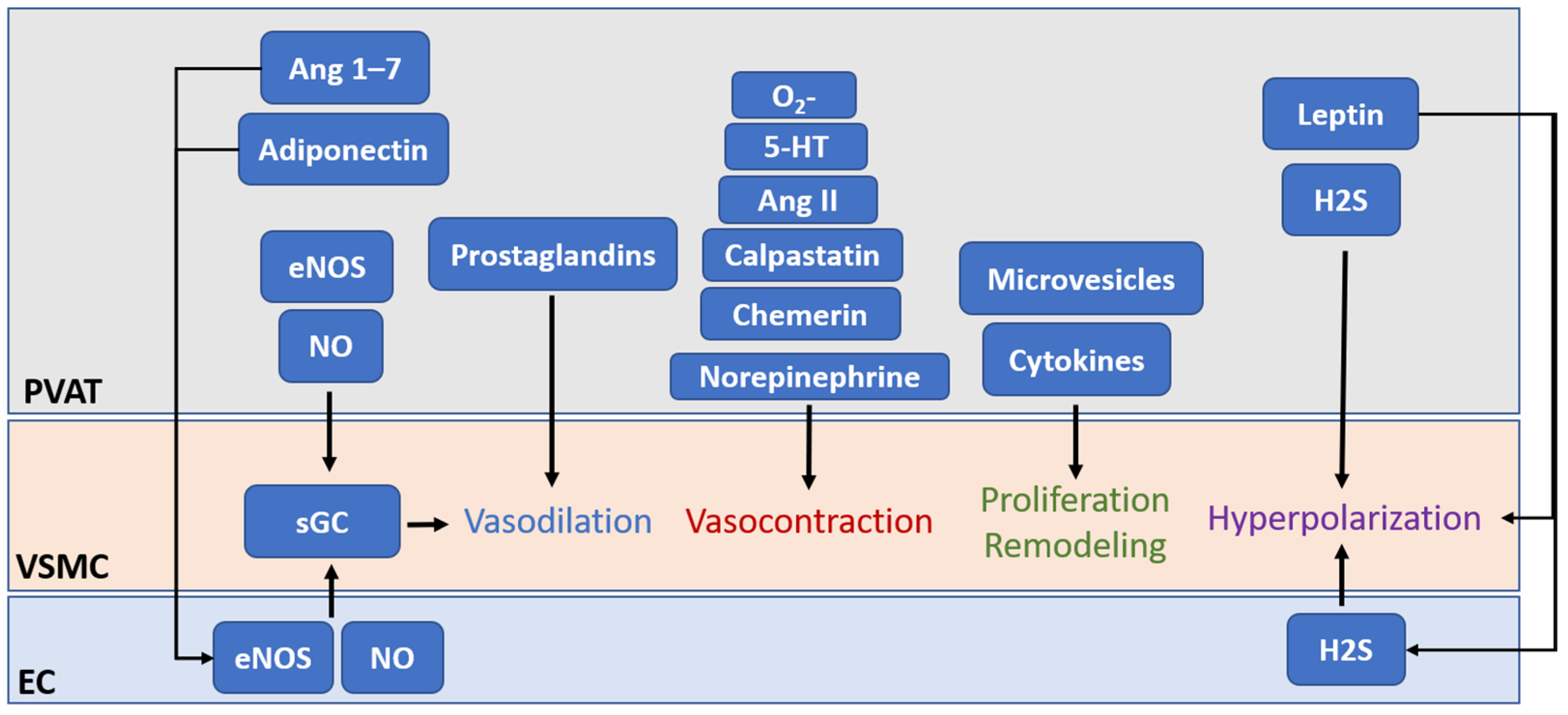
| PVAT-Derived Factors | Effects | References |
|---|---|---|
| Adiponectin | Relaxation | [40][24] |
| Angiotensin (Ang) 1–7 | Relaxation | [46][30] |
| Angiotensin II (Ang II) | Contraction | [14,56,57][41][42][43] |
| Calpastatin | Contraction | [51][35] |
| Chemerin | Contraction | [50,54][34][39] |
| Hydrogen peroxide (H2O2) | Relaxation | [42,55][26][40] |
| Hydrogen sulphide (H2S) | Contraction | [41][25] |
| Relaxation | [56,58][41][44] | |
| Leptin | Relaxation | [57,59][43][45] |
| Contraction | [51,60][35][46] | |
| Nitric oxide (NO) | Relaxation | [45][29] |
| Norepinephrine (NE) | Contraction | [52][36] |
| Prostanoids | ||
| -Prostaglandins | Contraction | [44,61][28][47] |
| -Prostacyclin | Relaxation | [22][48] |
| -Thromboxane | Contraction | [61][47] |
| Superoxide | Contraction | [53][37] |
| 5-hydroxytryptamine (5-HT) | Contraction | [49][33] |
3. Current Proves of eNOS in PVAT
There are currently three isoforms of NO synthase (NOS), which is named by the cell types where they are first identified: neuronal NOS (nNOS or NOS1), inducible NOS (iNOS or NOS2), and eNOS (or NOS3) [77][59]. Vascular nNOS is expressed in perivascular nerve fibers and in the vascular wall, while the expression of iNOS is induced under conditions of inflammation and sepsis [77][59]. eNOS is primarily expressed, but not exclusively, in endothelial cells. All three isoforms of NO synthase catalyze the production of NO from L-arginine [80][60]. Under physiological conditions, eNOS is the main vascular source of NO, modulates vascular functions and confers protection against cardiovascular diseases. In recent years, eNOS expression in other cell types has been demonstrated in vitro and in vivo. Indeed, eNOS expression has been found in dendrite cells [78][61], red blood cells [81][62], hepatocytes [80][60], as well as in adipocytes [6]. While the expression of iNOS in PVAT is only induced in pathological conditions [82][63], and the expression of nNOS in PVAT is controversial [83][64], the expression of eNOS in thoracic aortic PVAT has recently been demonstrated by various groups. Gene and protein expressions of eNOS in PVAT have been detected [6,84][6][65]. Using immunohistochemistry, eNOS can be stained in both adipocytes and endothelial cells of the capillaries and vasa vasorum in aortic PVAT [6,85][6][66]. Of the three isoforms of NOS, immunostaining of eNOS is the most abundant in PVAT of the saphenous vein, and eNOS activity is comparable in PVAT and the adherent vein [85][66]. In addition, in situ NO production in PVAT adipocytes can be directly detected by fluorescence imaging [13,86][67][68]. There is a high histological discrepancy of eNOS expression among the anatomical localizations of PVAT. Abdominal PVAT has been shown to have a lower eNOS expression compared with thoracic PVAT, while the eNOS expression remains the same along the vessel walls [13][67]. Indeed, unpublished data from ouresearchers' laboratory suggests a similar eNOS expression level of mesenteric PVAT and thoracic PVAT. Nevertheless, due to the highly heterogenous origins and compositions of different PVATs, detailed investigations of specific cell types that express eNOS in different PVATs is crucial.4. What Are the Functions of eNOS in PVAT?
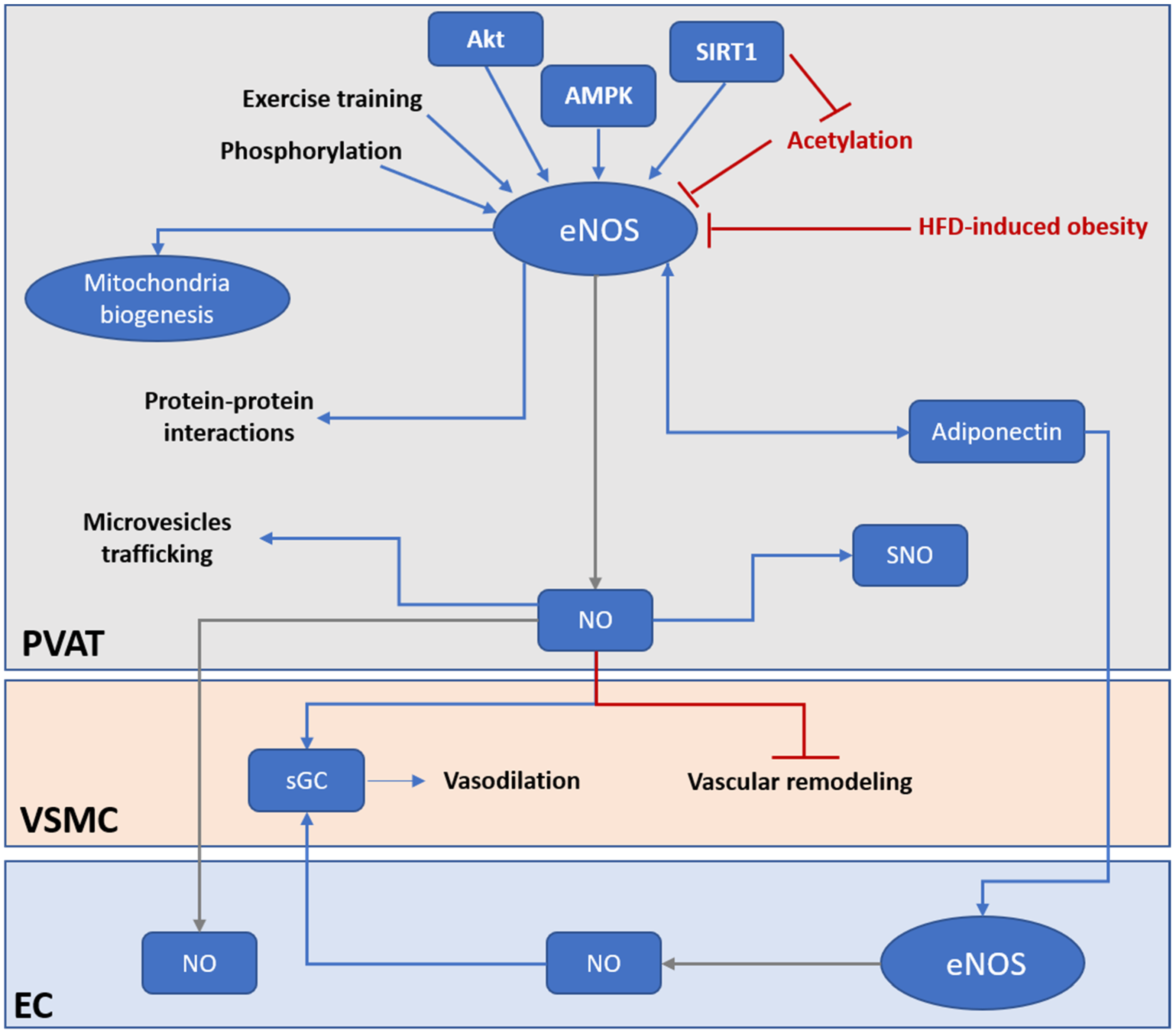
Unfortunately, due to the lack of PVAT-specific eNOS knockout animal models, the exact functions of eNOS in PVAT is relatively unclear. Most of the current knowledge about PVAT eNOS is based on evidence from studies using global eNOS knockout mice or mice with pathological conditions that leads to downregulation of PVAT eNOS. Here, wresearchers summarize current understanding of potential eNOS functions in PVAT under both basal and pathological conditions. The first and most important function of eNOS in PVAT is, of course, to generate vasoactive NO. Previous studies with animal models have demonstrated that PVAT plays a crucial role in vascular NO production [1,6,29][1][6][12]. PVAT-derived NO can diffuse into the adjacent VSMC, stimulating soluble guanylate cyclase (sGC) and increasing the cyclic guanosine monophosphate (cGMP) level, which leads to the phosphorylation of large-conductance calcium-activated potassium channels in VSMC via protein kinase G, resulting in hyperpolarization and vascular relaxation [87,88][69][70]. In small arteries isolated from visceral fat of healthy individuals, basal vascular NO production is reduced after PVAT removal, which leads to an attenuated contractile response to L-NAME [89][71]. In PVAT-adhered, endothelium-denuded rat mesenteric arteries, inhibition of eNOS significantly enhances NE-induced contraction, indicating that eNOS in PVAT contributes to the vascular NO production, while the anticontractile effect of PVAT is, at least partly, independent of the endothelium [33,90][17][72]. In low-density lipoprotein receptor (Ldlr) knockout mice, the thoracic aortic PVAT shows significant upregulation of eNOS expression and NO production, which protects against impaired vasorelaxation to acetylcholine and insulin [84][65]. In a very recent clinical study, the authors demonstrated PVAT as a predominant source of NO in human vasculature in a no-touch saphenous vein grafts (NT-SVGs) coronary artery bypass model [91][73]. The study showed that PVAT, via NO production from eNOS, can induce vasorelaxation even in endothelium-denuded SVG. The above evidence suggests the protective role of PVAT eNOS in improving endothelial functions. Nevertheless, currently, there is a lack of detailed studies that are designed to compare the NO production and eNOS function among vascular components, such as the endothelium and PVAT. In addition to direct modulation of vasodilation, PVAT-derived NO released toward the vascular lumen is a potent inhibitor of platelet aggregation and leukocyte adhesion [92][74]. PVAT has been shown to play a role in the inhibition of DNA synthesis, mitogenesis, and proliferation of VSMCs [93][75]. The inhibition of platelet aggregation and adhesion also protects VSMCs from exposure to platelet-derived growth factors. These confer the ability of PVAT to protect against the onset of atherogenesis and vascular remodeling in the adherent vessels. However, there is a lack of direct evidence of how PVAT eNOS and PVAT-derived NO act on atherogenesis and vascular remodeling. Another important function of PVAT eNOS is to stimulate the expression of adiponectin, which is an important adipokine that contributes to vasodilation regulation, anti-inflammation, and inhibition of VSMCs proliferation and migration [36,94][20][76]. eNOS has been shown to regulate adiponectin synthesis in adipocytes by increasing mitochondrial biogenesis and enhancing mitochondrial function [95][77]. PVAT-derived adiponectin may regulate endothelial functions, partly by enhancing eNOS phosphorylation in the endothelium [96][78]. Indeed, the function of PVAT is determined by the browning and inflammation status. Mitochondrial biogenesis is involved in the browning of adipocytes [97][79]. Fitzgibbons et al. proposed that promoting the browning of PVAT might confer a protective effect to attenuate the development of vascular diseases [11][80]. PVAT eNOS may have a vital role in the mitochondrial biogenesis and browning of PVAT [98][81]. However, the detailed mechanisms underlying browning or thermogenesis of PVAT are barely known. Apart from the functions of PVAT eNOS and NO mentioned above, NO is also known as an endogenously produced signaling molecule that regulates gene expression and cell phenotypes [99][82]. Currently, NO is known to regulate gene expression either by direct interaction with transcription factors or by post-translational modification of proteins. NO may mediate the transcriptional regulation of histone-modifying enzymes and modulate the activities and cellular localizations of transcription factors through the formation of S-nitrosothiols or iron nitrosyl complexes [100][83]. Additionally, NO may alter the cellular methylation, acetylation, phosphorylation, ubiquitylation, or sumoylation profiles of proteins and histones by modifying these enzymes [101][84]. Recent evidence has revealed the presence of S-nitrosylated (SNO) proteins in abdominal aortic PVAT [102][85]. For example, a reduced NO level results in the activation and cellular release of tissue transglutaminase (TG2), which is involved in vascular fibrosis and remodeling [103,104][86][87]. Normally, TG2 can be S-nitrosylated by NO, and is retained within the cytosolic compartment. Reduced bioavailability of NO leads to reduction of TG2 S-nitrosylation, which facilities its translocation to the extracellular compartment where it can induce crosslinking of extracellular matrix proteins and promote fibrosis [105][88]. Nevertheless, the complete nitrosylation profile of PVAT and vascular wall remains unclear. Identification of these SNO proteins could greatly enhance ouresearchers' understanding of the detailed function of PVAT eNOS and its derived NO. A recent study has revealed the reciprocal regulation of eNOS and β-catenin [106][89]. eNOS and β-catenin are interactive partners. β-catenin is a membrane protein known to bind with eNOS to promote eNOS phosphorylation and activation, while this interaction facilitates the translocation of β-catenin to the nucleus and activates downstream gene transcription [106][89]. This suggests a potential role of eNOS as a ‘carrier’ protein to facilitate gene expression independent of NO production. In addition, another cobinding protein and negative regulator of eNOS, Cav-1, is expressed in both endothelial cells and adipocytes [107][90]. Cav-1 can regulate eNOS functions in PVAT [108][91], whereas eNOS-derived NO has been shown to promote caveolae trafficking [109][92]. These suggest that protein–protein interaction of eNOS may play a critical role in PVAT functions, such as the secretion of miRNA-encapsulated microvesicles.5. What Are the Functions of eNOS in PVAT?
6. PVAT eNOS under Pathological Conditions
7. Pharmacological Targeting of PVAT eNOS
8. Conclusions and Future Directions
- o
-
What is the exact of role of PVAT eNOS in PVAT functions?
- o
-
What are the exact expression levels of eNOS in different regions of PVAT and/or in different cells in PVAT?
- o
-
What is the relative contribution of endothelial eNOS and PVAT eNOS to vascular function under physiological and pathological conditions?
- o
-
Are there any specific functions of eNOS in PVAT but not in endothelial cells?
Author Contributions
A.W.C.M. wrote the initial draft of the manuscript. Y.Z., N.X. and H.L. critically reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript.Funding
Original works from the authors’ laboratory contributing to this review were supported by grants LI-1042/1-1, LI-1042/3-1, LI-1042/5-1, and XI 139/2-1 from the Deutsche Forschungsgemeinschaft (DFG), Bonn, Germany. H.L. and N.X. were supported by a research grant from the Boehringer Ingelheim Foundation for the collaborative research consortium “Novel and neglected cardiovascular risk factors: molecular mechanisms and therapeutic implications”.Institutional Review Board Statement
Not applicable.Informed Consent Statement
Not applicable.Data Availability Statement
Not applicable.Conflicts of Interest
The authors declare no conflict of interest.
References
- Xia, N.; Li, H. The role of perivascular adipose tissue in obesity-induced vascular dysfunction. Br. J. Pharmacol. 2017, 174, 3425–3442.
- Soltis, E.E.; Cassis, L.A. Influence of Perivascular Adipose Tissue on Rat Aortic Smooth Muscle Responsiveness. Clin. Exp. Hypertens. Part A Theory Pract. 1991, 13, 277–296.
- Gao, Y.-J.; Zeng, Z.-H.; Teoh, K.; Sharma, A.M.; Abouzahr, L.; Cybulsky, I.; Lamy, A.; Semelhago, L.; Lee, R.M. Perivascular adipose tissue modulates vascular function in the human internal thoracic artery. J. Thorac. Cardiovasc. Surg. 2005, 130, 1130–1136.
- Greenstein, A.S.; Khavandi, K.; Withers, S.B.; Sonoyama, K.; Clancy, O.; Jeziorska, M.; Laing, I.; Yates, A.P.; Pemberton, P.W.; Malik, R.A.; et al. Local Inflammation and Hypoxia Abolish the Protective Anticontractile Properties of Perivascular Fat in Obese Patients. Circulation 2009, 119, 1661–1670.
- Loesch, A.; Dashwood, M.R. Saphenous Vein Vasa Vasorum as a Potential Target for Perivascular Fat-Derived Factors. Braz. J. Cardiovasc. Surg. 2020, 35, 964–969.
- Xia, N.; Horke, S.; Habermeier, A.; Closs, E.I.; Reifenberg, G.; Gericke, A.; Mikhed, Y.; Münzel, T.; Daiber, A.; Förstermann, U.; et al. Uncoupling of Endothelial Nitric Oxide Synthase in Perivascular Adipose Tissue of Diet-Induced Obese Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 78–85.
- Chau, Y.-Y.; Bandiera, R.; Serrels, A.; Estrada, O.M.M.; Qing, W.; Lee, M.; Slight, J.; Thornburn, A.; Berry, R.; McHaffie, S.; et al. Visceral and subcutaneous fat have different origins and evidence supports a mesothelial source. Nat. Cell Biol. 2014, 16, 367–375.
- Lehr, S.; Hartwig, S.; Lamers, D.; Famulla, S.; Müller, S.; Hanisch, F.-G.; Cuvelier, C.; Ruige, J.; Eckardt, K.; Ouwens, D.M.; et al. Identification and Validation of Novel Adipokines Released from Primary Human Adipocytes. Mol. Cell. Proteom. 2012, 11, M111.010504.
- Löhn, M.; Dubrovska, G.; Lauterbach, B.; Luft, F.C.; Gollasch, M.; Sharma, A.M. Periadventitial fat releases a vascular relaxing factor. FASEB J. 2002, 16, 1057–1063.
- Xia, N.; Förstermann, U.; Li, H. Effects of resveratrol on eNOS in the endothelium and the perivascular adipose tissue. Ann. New York Acad. Sci. 2017, 1403, 132–141.
- Man, A.W.C.; Zhou, Y.; Xia, N.; Li, H. Perivascular Adipose Tissue as a Target for Antioxidant Therapy for Cardiovascular Complications. Antioxidants 2020, 9, 574.
- Chang, L.; Garcia-Barrio, M.T.; Chen, Y.E. Perivascular Adipose Tissue Regulates Vascular Function by Targeting Vascular Smooth Muscle Cells. Arter. Thromb. Vasc. Biol. 2020, 40, 1094–1109.
- Kim, H.W.; Shi, H.; Winkler, M.A.; Lee, R.; Weintraub, N.L. Perivascular Adipose Tissue and Vascular Perturbation/Atherosclerosis. Arter. Thromb. Vasc. Biol. 2020, 40, 2569–2576.
- Stanek, A.; Brożyna-Tkaczyk, K.; Myśliński, W. The Role of Obesity-Induced Perivascular Adipose Tissue (PVAT) Dysfunction in Vascular Homeostasis. Nutrients 2021, 13, 3843.
- Brown, N.K.; Zhou, Z.; Zhang, J.; Zeng, R.; Wu, J.; Eitzman, D.T.; Chen, Y.E.; Chang, L. Perivascular Adipose Tissue in Vascular Function and Disease. Arter. Thromb. Vasc. Biol. 2014, 34, 1621–1630.
- Omar, A.; Chatterjee, T.K.; Tang, Y.; Hui, D.Y.; Weintraub, N.L. Proinflammatory Phenotype of Perivascular Adipocytes. Arter. Thromb. Vasc. Biol. 2014, 34, 1631–1636.
- Bussey, C.E.; Withers, S.B.; Aldous, R.G.; Edwards, G.; Heagerty, A.M. Obesity-Related Perivascular Adipose Tissue Damage Is Reversed by Sustained Weight Loss in the Rat. Arter. Thromb. Vasc. Biol. 2016, 36, 1377–1385.
- Ketonen, J.; Shi, J.; Martonen, E.; Mervaala, E. Periadventitial Adipose Tissue Promotes Endothelial Dysfunction via Oxidative Stress in Diet-Induced Obese C57Bl/6 Mice. Circ. J. 2010, 74, 1479–1487.
- Xia, N.; Reifenberg, G.; Schirra, C.; Li, H. The Involvement of Sirtuin 1 Dysfunction in High-Fat Diet-Induced Vascular Dysfunction in Mice. Antioxidants 2022, 11, 541.
- Sowka, A.; Dobrzyn, P. Role of Perivascular Adipose Tissue-Derived Adiponectin in Vascular Homeostasis. Cells 2021, 10, 1485.
- Takaoka, M.; Nagata, D.; Kihara, S.; Shimomura, I.; Kimura, Y.; Tabata, Y.; Saito, Y.; Nagai, R.; Sata, M. Periadventitial Adipose Tissue Plays a Critical Role in Vascular Remodeling. Circ. Res. 2009, 105, 906–911.
- Chang, L.; Zhao, X.; Garcia-Barrio, M.; Zhang, J.; Chen, Y.E. MitoNEET in Perivascular Adipose Tissue Prevents Arterial Stiffness in Aging Mice. Cardiovasc. Drugs Ther. 2018, 32, 531–539.
- Chang, L.; Milton, H.; Eitzman, D.T.; Chen, Y.E. Paradoxical Roles of Perivascular Adipose Tissue in Atherosclerosis and Hypertension. Circ. J. 2013, 77, 11–18.
- Fésüs, G.; Dubrovska, G.; Gorzelniak, K.; Kluge, R.; Huang, Y.; Luft, F.C.; Gollasch, M. Adiponectin is a novel humoral vasodilator. Cardiovasc. Res. 2007, 75, 719–727.
- Wójcicka, G.; Jamroz-Wiśniewska, A.; Atanasova, P.; Chaldakov, G.N.; Chylińska-Kula, B.; Bełtowski, J. Differential effects of statins on endogenous H2S formation in perivascular adipose tissue. Pharmacol. Res. 2011, 63, 68–76.
- Zaborska, K.E.; Wareing, M.; Austin, C. Comparisons between perivascular adipose tissue and the endothelium in their modulation of vascular tone. Br. J. Pharmacol. 2017, 174, 3388–3397.
- Awata, W.M.; Gonzaga, N.A.; Borges, V.F.; Silva, C.B.; Tanus-Santos, J.E.; Cunha, F.Q.; Tirapelli, C.R. Perivascular adipose tissue contributes to lethal sepsis-induced vasoplegia in rats. Eur. J. Pharmacol. 2019, 863, 172706.
- Ozen, G.; Topal, G.; Gomez, I.; Ghorreshi, A.; Boukais, K.; Benyahia, C.; Kanyinda, L.; Longrois, D.; Teskin, O.; Uydes-Dogan, B.S.; et al. Control of human vascular tone by prostanoids derived from perivascular adipose tissue. Prostaglandins Other Lipid Mediat. 2013, 107, 13–17.
- Gao, Y.-J.; Lu, C.; Su, L.-Y.; Sharma, A.M.; Lee, R.M.K.W. Modulation of vascular function by perivascular adipose tissue: The role of endothelium and hydrogen peroxide. J. Cereb. Blood Flow Metab. 2007, 151, 323–331.
- Lee, R.M.K.W.; Lu, C.; Su, L.-Y.; Gao, Y.-J. Endothelium-dependent relaxation factor released by perivascular adipose tissue. J. Hypertens. 2009, 27, 782–790.
- Chang, L.; Xiong, W.; Zhao, X.; Fan, Y.; Guo, Y.; Garcia-Barrio, M.; Zhang, J.; Jiang, Z.; Lin, J.D.; Chen, Y.E. Bmal1 in Perivascular Adipose Tissue Regulates Resting-Phase Blood Pressure Through Transcriptional Regulation of Angiotensinogen. Circulation 2018, 138, 67–79.
- Alberti, K.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.-C.; James, W.P.T.; Loria, C.M.; Smith, S.C., Jr. Harmonizing the metabolic syndrome: A joint interim statement of the international diabetes federation task force on epidemiology and prevention; national heart, lung, and blood institute; American heart association; world heart federation; international atherosclerosis society; and international association for the study of obesity. Circulation 2009, 120, 1640–1645.
- Kumar, R.K.; Darios, E.S.; Burnett, R.; Thompson, J.M.; Watts, S.W. Fenfluramine-induced PVAT-dependent contraction depends on norepinephrine and not serotonin. Pharmacol. Res. 2018, 140, 43–49.
- Watts, S.W.; Dorrance, A.M.; Penfold, M.E.; Rourke, J.L.; Sinal, C.J.; Seitz, B.; Sullivan, T.J.; Charvat, T.T.; Thompson, J.M.; Burnett, R.; et al. Chemerin Connects Fat to Arterial Contraction. Arter. Thromb. Vasc. Biol. 2013, 33, 1320–1328.
- Owen, M.K.; Witzmann, F.A.; McKenney, M.L.; Lai, X.; Berwick, Z.C.; Moberly, S.P.; Alloosh, M.; Sturek, M.; Tune, J.D. Perivascular adipose tissue potentiates contraction of coronary vascular smooth muscle: Influence of obesity. Circulation 2013, 128, 9–18.
- Ayala-Lopez, N.; Martini, M.; Jackson, W.F.; Darios, E.; Burnett, R.; Seitz, B.; Fink, G.D.; Watts, S.W. Perivascular adipose tissue contains functional catecholamines. Pharmacol. Res. Perspect. 2014, 2, e00041.
- Gao, Y.-J.; Takemori, K.; Su, L.-Y.; An, W.-S.; Lu, C.; Sharma, A.M.; Lee, R.M. Perivascular adipose tissue promotes vasoconstriction: The role of superoxide anion. Cardiovasc. Res. 2006, 71, 363–373.
- Gil-Ortega, M.; Somoza, B.; Huang, Y.; Gollasch, M.; Fernández-Alfonso, M.S. Regional differences in perivascular adipose tissue impacting vascular homeostasis. Trends Endocrinol. Metab. 2015, 26, 367–375.
- Darios, E.S.; Winner, B.M.; Charvat, T.; Krasinksi, A.; Punna, S.; Watts, S.W. The adipokine chemerin amplifies electrical field-stimulated contraction in the isolated rat superior mesenteric artery. Am. J. Physiol. Circ. Physiol. 2016, 311, H498–H507.
- Costa, R.M.; Filgueira, F.P.; Tostes, R.C.; Carvalho, M.H.C.; Akamine, E.H.; Lobato, N.S. H2O2 generated from mitochondrial electron transport chain in thoracic perivascular adipose tissue is crucial for modulation of vascular smooth muscle contraction. Vasc. Pharmacol. 2016, 84, 28–37.
- Cacanyiova, S.; Majzunova, M.; Golas, S.; Berenyiova, A. The role of perivascular adipose tissue and endogenous hydrogen sulfide in vasoactive responses of isolated mesenteric arteries in normotensive and spontaneously hypertensive rats. J. Physiol. Pharmacol. 2019, 70, 295–306.
- Gálvez-Prieto, B.; Bolbrinker, J.; Stucchi, P.; de Las Heras, A.I.; Merino, B.; Arribas, S.; Ruiz-Gayo, M.; Huber, M.; Wehland, M.; Kreutz, R.; et al. Comparative expression analysis of the renin–angiotensin system components between white and brown perivascular adipose tissue. J. Endocrinol. 2008, 197, 55–64.
- Gálvez-Prieto, B.; Somoza, B.; Gil-Ortega, M.; García-Prieto, C.F.; de las Heras, A.I.; González, M.C.; Arribas, S.; Aranguez, I.; Bolbrinker, J.; Kreutz, R.; et al. Anticontractile Effect of Perivascular Adipose Tissue and Leptin are Reduced in Hypertension. Front. Pharmacol. 2012, 3, 103.
- Fang, L.; Zhao, J.; Chen, Y.; Ma, T.; Xu, G.; Tang, C.; Liu, X.; Geng, B. Hydrogen sulfide derived from periadventitial adipose tissue is a vasodilator. J. Hypertens. 2009, 27, 2174–2185.
- Payne, G.A.; Borbouse, L.; Kumar, S.; Neeb, Z.; Alloosh, M.; Sturek, M.; Tune, J.D. Epicardial Perivascular Adipose-Derived Leptin Exacerbates Coronary Endothelial Dysfunction in Metabolic Syndrome via a Protein Kinase C-β Pathway. Arter. Thromb. Vasc. Biol. 2010, 30, 1711–1717.
- Noblet, J.N.; Goodwill, A.; Sassoon, D.; Kiel, A.; Tune, J.D. Leptin augments coronary vasoconstriction and smooth muscle proliferation via a Rho-kinase-dependent pathway. Basic Res. Cardiol. 2016, 111, 25.
- Mendizábal, Y.; Llorens, S.; Nava, E. Vasoactive effects of prostaglandins from the perivascular fat of mesenteric resistance arteries in WKY and SHROB rats. Life Sci. 2013, 93, 1023–1032.
- Chang, L.; Villacorta, L.; Li, R.; Hamblin, M.; Xu, W.; Dou, C.; Zhang, J.; Wu, J.; Zeng, R.; Chen, Y.E. Loss of Perivascular Adipose Tissue on Peroxisome Proliferator–Activated Receptor-γ Deletion in Smooth Muscle Cells Impairs Intravascular Thermoregulation and Enhances Atherosclerosis. Circulation 2012, 126, 1067–1078.
- Ayala-Lopez, N.; Thompson, J.M.; Watts, S.W. Perivascular Adipose Tissue’s Impact on Norepinephrine-Induced Contraction of Mesenteric Resistance Arteries. Front. Physiol. 2017, 8, 37.
- Saxton, S.N.; Ryding, K.E.; Aldous, R.G.; Withers, S.B.; Ohanian, J.; Heagerty, A.M. Role of Sympathetic Nerves and Adipocyte Catecholamine Uptake in the Vasorelaxant Function of Perivascular Adipose Tissue. Arter. Thromb. Vasc. Biol. 2018, 38, 880–891.
- Jackson, W.F. Myogenic Tone in Peripheral Resistance Arteries and Arterioles: The Pressure Is On! Front. Physiol. 2021, 12, 699517.
- Schmidt, K.; De Wit, C. Endothelium-Derived Hyperpolarizing Factor and Myoendothelial Coupling: The In Vivo Perspective. Front. Physiol. 2020, 11, 602930.
- Watts, S.W.; Flood, E.D.; Garver, H.; Fink, G.D.; Roccabianca, S. A New Function for Perivascular Adipose Tissue (PVAT): Assistance of Arterial Stress Relaxation. Sci. Rep. 2020, 10, 1807.
- Miron, T.R.; Flood, E.D.; Tykocki, N.R.; Thompson, J.M.; Watts, S.W. Identification of Piezo1 channels in perivascular adipose tissue (PVAT) and their potential role in vascular function. Pharmacol. Res. 2021, 175, 105995.
- Ahmadieh, S.; Kim, H.W.; Weintraub, N.L. Potential role of perivascular adipose tissue in modulating atherosclerosis. Clin. Sci. 2020, 134, 3–13.
- Hu, H.; Garcia-Barrio, M.; Jiang, Z.-S.; Chen, Y.E.; Chang, L. Roles of Perivascular Adipose Tissue in Hypertension and Atherosclerosis. Antioxidants Redox Signal. 2021, 34, 736–749.
- Farias-Itao, D.S.; Pasqualucci, C.A.; de Andrade, R.A.; da Silva, L.F.F.; Yahagi-Estevam, M.; Lage, S.H.G.; Leite, R.E.P.; Campo, A.B.; Suemoto, C.K. Macrophage Polarization in the Perivascular Fat Was Associated with Coronary Atherosclerosis. J. Am. Hear. Assoc. 2022, 11, e023274.
- Irie, D.; Kawahito, H.; Wakana, N.; Kato, T.; Kishida, S.; Kikai, M.; Ogata, T.; Ikeda, K.; Ueyama, T.; Matoba, S.; et al. Transplantation of periaortic adipose tissue from angiotensin receptor blocker-treated mice markedly ameliorates atherosclerosis development in apoE–/– mice. J. Renin-Angiotensin-Aldosterone Syst. 2014, 16, 67–78.
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837.
- Bendall, J.K.; Alp, N.J.; Warrick, N.; Cai, S.; Adlam, D.; Rockett, K.; Yokoyama, M.; Kawashima, S.; Channon, K.M. Stoichiometric Relationships Between Endothelial Tetrahydrobiopterin, Endothelial NO Synthase (eNOS) Activity, and eNOS Coupling in Vivo. Circ. Res. 2005, 97, 864–871.
- Caviedes, A.; Varas-Godoy, M.; Lafourcade, C.; Sandoval, S.; Bravo-Alegria, J.; Kaehne, T.; Massmann, A.; Figueroa, J.P.; Nualart, F.; Wyneken, U. Endothelial Nitric Oxide Synthase Is Present in Dendritic Spines of Neurons in Primary Cultures. Front. Cell. Neurosci. 2017, 11, 180.
- Leo, F.; Suvorava, T.; Heuser, S.K.; Li, J.; LoBue, A.; Barbarino, F.; Piragine, E.; Schneckmann, R.; Hutzler, B.; Good, M.E.; et al. Red Blood Cell and Endothelial eNOS Independently Regulate Circulating Nitric Oxide Metabolites and Blood Pressure. Circulation 2021, 144, 870–889.
- Nakladal, D.; Sijbesma, J.; Visser, L.; Tietge, U.; Slart, R.; Deelman, L.; Henning, R.; Hillebrands, J.; Buikema, H. Perivascular adipose tissue-derived nitric oxide compensates endothelial dysfunction in aged pre-atherosclerotic apolipoprotein E-deficient rats. Vasc. Pharmacol. 2021, 142, 106945.
- Nava, E.; Llorens, S. The Local Regulation of Vascular Function: From an Inside-Outside to an Outside-Inside Model. Front. Physiol. 2019, 10, 729.
- Baltieri, N.; Guizoni, D.M.; Victório, J.A.; Davel, A.P. Protective Role of Perivascular Adipose Tissue in Endothelial Dysfunction and Insulin-Induced Vasodilatation of Hypercholesterolemic LDL Receptor-Deficient Mice. Front. Physiol. 2018, 9, 229.
- Dashwood, M.R.; Dooley, A.; Shi-Wen, X.; Abraham, D.J.; Souza, D.S. Does Periadventitial Fat-Derived Nitric Oxide Play a Role in Improved Saphenous Vein Graft Patency in Patients Undergoing Coronary Artery Bypass Surgery? J. Vasc. Res. 2007, 44, 175–181.
- Victorio, J.A.; Fontes, M.T.; Rossoni, L.V.; Davel, A.P. Different Anti-Contractile Function and Nitric Oxide Production of Thoracic and Abdominal Perivascular Adipose Tissues. Front. Physiol. 2016, 7, 295.
- Gil-Ortega, M.; Stucchi, P.; Guzmán-Ruiz, R.; Cano, V.; Arribas, S.; González, M.C.; Ruiz-Gayo, M.; Fernández-Alfonso, M.S.; Somoza, B. Adaptative Nitric Oxide Overproduction in Perivascular Adipose Tissue during Early Diet-Induced Obesity. Endocrinology 2010, 151, 3299–3306.
- Förstermann, U.; Closs, E.I.; Pollock, J.S.; Nakane, M.; Schwarz, P.; Gath, I.; Kleinert, H. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension 1994, 23, 1121–1131.
- Weston, A.H.; Egner, I.; Dong, Y.; Porter, E.L.; Heagerty, A.M.; Edwards, G. Stimulated release of a hyperpolarizing factor (ADHF) from mesenteric artery perivascular adipose tissue: Involvement of myocyte BKCachannels and adiponectin. J. Cereb. Blood Flow Metab. 2013, 169, 1500–1509.
- Virdis, A.; Duranti, E.; Rossi, C.; Dell’Agnello, U.; Santini, E.; Anselmino, M.; Chiarugi, M.; Taddei, S.; Solini, A. Tumour necrosis factor-alpha participates on the endothelin-1/nitric oxide imbalance in small arteries from obese patients: Role of perivascular adipose tissue. Eur. Hear. J. 2014, 36, 784–794.
- Aghamohammadzadeh, R.; Unwin, R.D.; Greenstein, A.S.; Heagerty, A.M. Effects of Obesity on Perivascular Adipose Tissue Vasorelaxant Function: Nitric Oxide, Inflammation and Elevated Systemic Blood Pressure. J. Vasc. Res. 2015, 52, 299–305.
- Saito, T.; Kurazumi, H.; Suzuki, R.; Matsunaga, K.; Tsubone, S.; Lv, B.; Kobayashi, S.; Nagase, T.; Mizoguchi, T.; Samura, M.; et al. Perivascular Adipose Tissue Is a Major Source of Nitric Oxide in Saphenous Vein Grafts Harvested via the No-Touch Technique. J. Am. Hear. Assoc. 2022, 11, e020637.
- Qi, X.-Y.; Qu, S.-L.; Xiong, W.-H.; Rom, O.; Chang, L.; Jiang, Z.-S. Perivascular adipose tissue (PVAT) in atherosclerosis: A double-edged sword. Cardiovasc. Diabetol. 2018, 17, 134.
- Barandier, C.; Montani, J.-P.; Yang, Z. Mature adipocytes and perivascular adipose tissue stimulate vascular smooth muscle cell proliferation: Effects of aging and obesity. Am. J. Physiol. Circ. Physiol. 2005, 289, H1807–H1813.
- Matsuzawa, Y. Adiponectin: A Key Player in Obesity Related Disorders. Curr. Pharm. Des. 2010, 16, 1896–1901.
- Koh, E.H.; Kim, M.; Ranjan, K.C.; Kim, H.S.; Park, H.-S.; Oh, K.S.; Park, I.-S.; Lee, W.J.; Kim, M.-S.; Park, J.-Y.; et al. eNOS plays a major role in adiponectin synthesis in adipocytes. Am. J. Physiol. Metab. 2010, 298, E846–E853.
- Sena, C.M.; Pereira, A.; Fernandes, R.; Letra, L.; Seiça, R.M. Adiponectin improves endothelial function in mesenteric arteries of rats fed a high-fat diet: Role of perivascular adipose tissue. J. Cereb. Blood Flow Metab. 2017, 174, 3514–3526.
- Lemecha, M.; Morino, K.; Imamura, T.; Iwasaki, H.; Ohashi, N.; Ida, S.; Sato, D.; Sekine, O.; Ugi, S.; Maegawa, H. MiR-494-3p regulates mitochondrial biogenesis and thermogenesis through PGC1-α signalling in beige adipocytes. Sci. Rep. 2018, 8, 15096.
- Fitzgibbons, T.P.; Kogan, S.; Aouadi, M.; Hendricks, G.M.; Straubhaar, J.; Czech, M.P. Similarity of mouse perivascular and brown adipose tissues and their resistance to diet-induced inflammation. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1425–H1437.
- Kong, L.-R.; Zhou, Y.-P.; Chen, D.-R.; Ruan, C.-C.; Gao, P.-J. Decrease of Perivascular Adipose Tissue Browning Is Associated With Vascular Dysfunction in Spontaneous Hypertensive Rats During Aging. Front. Physiol. 2018, 9, 400.
- Hofseth, L.J.; Robles, A.I.; Espey, M.G.; Harris, C.C. Nitric Oxide Is a Signaling Molecule that Regulates Gene Expression. Methods Enzymol. 2005, 396, 326–340.
- Socco, S.; Bovee, R.C.; Palczewski, M.B.; Hickok, J.R.; Thomas, D.D. Epigenetics: The third pillar of nitric oxide signaling. Pharmacol. Res. 2017, 121, 52–58.
- Vasudevan, D.; Hickok, J.R.; Bovee, R.C.; Pham, V.; Mantell, L.L.; Bahroos, N.; Kanabar, P.; Cao, X.-J.; Maienschein-Cline, M.; Garcia, B.A.; et al. Nitric Oxide Regulates Gene Expression in Cancers by Controlling Histone Posttranslational Modifications. Cancer Res. 2015, 75, 5299–5308.
- Barp, C.G.; Benedet, P.O.; Assreuy, J. Perivascular adipose tissue phenotype and sepsis vascular dysfunction: Differential contribution of NO, ROS and beta 3-adrenergic receptor. Life Sci. 2020, 254, 117819.
- Jia, G.; Aroor, A.R.; Martinez-Lemus, L.A.; Sowers, J.R. Overnutrition, mTOR signaling, and cardiovascular diseases. Am. J. Physiol. Integr. Comp. Physiol. 2014, 307, R1198–R1206.
- Jia, G.; DeMarco, V.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2015, 12, 144–153.
- Jia, G.; Habibi, J.; Aroor, A.R.; Martinez-Lemus, L.A.; DeMarco, V.G.; Ramirez-Perez, F.I.; Sun, Z.; Hayden, M.R.; Meininger, G.A.; Mueller, K.B.; et al. Endothelial Mineralocorticoid Receptor Mediates Diet-Induced Aortic Stiffness in Females. Circ. Res. 2016, 118, 935–943.
- Tajadura, V.; Hansen, M.H.; Smith, J.; Charles, H.; Rickman, M.; Farrell-Dillon, K.; Claro, V.; Warboys, C.; Ferro, A. β-catenin promotes endothelial survival by regulating eNOS activity and flow-dependent anti-apoptotic gene expression. Cell Death Dis. 2020, 11, 493.
- Cohen, A.W.; Hnasko, R.; Schubert, W.; Lisanti, M.P. Role of Caveolae and Caveolins in Health and Disease. Physiol. Rev. 2004, 84, 1341–1379.
- Lee, M.H.-H.; Chen, S.-J.; Tsao, C.-M.; Wu, C.-C. Perivascular Adipose Tissue Inhibits Endothelial Function of Rat Aortas via Caveolin-1. PLoS ONE 2014, 9, e99947.
- Chen, Z.; Oliveira, S.D.; Zimnicka, A.M.; Jiang, Y.; Sharma, T.; Chen, S.; Lazarov, O.; Bonini, M.G.; Haus, J.M.; Minshall, R.D. Reciprocal regulation of eNOS and caveolin-1 functions in endothelial cells. Mol. Biol. Cell 2018, 29, 1190–1202.
- Korda, M.; Kubant, R.; Patton, S.; Malinski, T. Leptin-induced endothelial dysfunction in obesity. Am. J. Physiol. Circ. Physiol. 2008, 295, H1514–H1521.
- Xia, N.; Weisenburger, S.; Koch, E.; Burkart, M.; Reifenberg, G.; Förstermann, U.; Li, H. Restoration of perivascular adipose tissue function in diet-induced obese mice without changing bodyweight. Br. J. Pharmacol. 2017, 174, 3443–3453.
- Gil-Ortega, M.; Condezo-Hoyos, L.; García-Prieto, C.F.; Arribas, S.M.; Gonzalez-Garcia, M.C.; Aranguez, I.; Ruiz-Gayo, M.; Somoza, B.; Fernandez-Alfonso, M.S. Imbalance between Pro and Anti-Oxidant Mechanisms in Perivascular Adipose Tissue Aggravates Long-Term High-Fat Diet-Derived Endothelial Dysfunction. PLoS ONE 2014, 9, e95312.
- Marchesi, C.; Ebrahimian, T.; Angulo, O.; Paradis, P.; Schiffrin, E.L. Endothelial Nitric Oxide Synthase Uncoupling and Perivascular Adipose Oxidative Stress and Inflammation Contribute to Vascular Dysfunction in a Rodent Model of Metabolic Syndrome. Hypertension 2009, 54, 1384–1392.
- Eyang, Z.; Eming, X.-F. Arginase: The Emerging Therapeutic Target for Vascular Oxidative Stress and Inflammation. Front. Immunol. 2013, 4, 149.
- Heiss, E.H.; Dirsch, V.M. Regulation of eNOS enzyme activity by posttranslational modification. Curr. Pharm. Des. 2014, 20, 3503–3513.
- Mattagajasingh, I.; Kim, C.-S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.-B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860.
- da Costa, R.M.; Silva, J.F.d.; Alves, J.V.; Dias, T.B.; Rassi, D.M.; Garcia, L.V.; Lobato, N.d.S.; Tostes, R.C. Increased O-GlcNAcylation of endothelial nitric oxide synthase compromises the anti-contractile properties of perivascular adipose tissue in metabolic syndrome. Front. Physiol. 2018, 9, 341.
- Yu, Y.; Rajapakse, A.G.; Montani, J.-P.; Yang, Z.; Ming, X.-F. p38 mitogen-activated protein kinase is involved in arginase-II-mediated eNOS-Uncoupling in Obesity. Cardiovasc. Diabetol. 2014, 13, 113.
- Flood, E.D.; Watts, S.W. Endogenous Chemerin from PVAT Amplifies Electrical Field-Stimulated Arterial Contraction: Use of the Chemerin Knockout Rat. Int. J. Mol. Sci. 2020, 21, 6392.
- Neves, K.B.; Lobato, N.S.; Lopes, R.A.M.; Filgueira, F.P.; Zanotto, C.Z.; Oliveira, A.M.; Tostes, R.C. Chemerin reduces vascular nitric oxide/cGMP signalling in rat aorta: A link to vascular dysfunction in obesity? Clin. Sci. 2014, 127, 111–122.
- Neves, K.B.; Cat, A.N.D.; Lopes, R.A.M.; Rios, F.J.; Anagnostopoulou, A.; Lobato, N.S.; de Oliveira, A.M.; Tostes, R.C.; Montezano, A.C.; Touyz, R.M. Chemerin Regulates Crosstalk Between Adipocytes and Vascular Cells Through Nox. Hypertension 2015, 66, 657–666.
- Miao, C.-Y.; Li, Z.-Y. The role of perivascular adipose tissue in vascular smooth muscle cell growth. J. Cereb. Blood Flow Metab. 2012, 165, 643–658.
- Gómez-Serrano, M.; Camafeita, E.; López, J.A.; Rubio, M.A.; Bretón, I.; García-Consuegra, I.; García-Santos, E.; Lago, J.; Sánchez-Pernaute, A.; Torres, A.; et al. Differential proteomic and oxidative profiles unveil dysfunctional protein import to adipocyte mitochondria in obesity-associated aging and diabetes. Redox Biol. 2016, 11, 415–428.
- Bailey-Downs, L.C.; Tucsek, Z.; Toth, P.; Sosnowska, D.; Gautam, T.; Sonntag, W.E.; Csiszar, A.; Ungvari, Z. Aging exacerbates obesity-induced oxidative stress and inflammation in perivascular adipose tissue in mice: A paracrine mechanism contributing to vascular redox dysregulation and inflammation. J. Gerontol. Ser. A Biomed. Sci. Med Sci. 2013, 68, 780–792.
- Guo, X.; Zhang, Y.; Zheng, L.; Zheng, C.; Song, J.; Zhang, Q.; Kang, B.; Liu, Z.; Jin, L.; Xing, R.; et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018, 24, 978–985.
- Yang, Y.-M.; Huang, A.; Kaley, G.; Sun, D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am. J. Physiol. Circ. Physiol. 2009, 297, H1829–H1836.
- Sousa, A.S.; Sponton, A.C.d.S.; Trifone, C.B.; Delbin, M.A. Aerobic exercise training prevents perivascular adipose tissue (PVAT)-induced endothelial dysfunction in thoracic aorta of obese mice. Front. Physiol. 2019, 10, 1009.
- You, T.; Arsenis, N.C.; Disanzo, B.L.; LaMonte, M.J. Effects of Exercise Training on Chronic Inflammation in Obesity. Sports Med. 2013, 43, 243–256.
- Gu, P.; Hui, H.; Vanhoutte, P.; Lam, K.; Xu, A. Deletion of SIRT1 in perivascular adipose tissue accelerates obesity-induced endothelial dysfunction. In Proceedings of the 1st ASCEPT-BPS Joint Scientific Meeting, Hong Kong, China, 19–21 May 2015.
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 Functionally Interacts with the Metabolic Regulator and Transcriptional Coactivator PGC-1α. J. Biol. Chem. 2005, 280, 16456–16460.
- Qiang, L.; Wang, H.; Farmer, S.R. Adiponectin Secretion Is Regulated by SIRT1 and the Endoplasmic Reticulum Oxidoreductase Ero1-Lα. Mol. Cell. Biol. 2007, 27, 4698–4707.
- Sun, Y.; Li, J.; Xiao, N.; Wang, M.; Kou, J.; Qi, L.; Huang, F.; Liu, B.; Liu, K. Pharmacological activation of AMPK ameliorates perivascular adipose/endothelial dysfunction in a manner interdependent on AMPK and SIRT1. Pharmacol. Res. 2014, 89, 19–28.
- Chen, Y.; Xu, X.; Zhang, Y.; Liu, K.; Huang, F.; Liu, B.; Kou, J. Diosgenin regulates adipokine expression in perivascular adipose tissue and ameliorates endothelial dysfunction via regulation of AMPK. J. Steroid Biochem. Mol. Biol. 2016, 155, 155–165.
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605.
- Bijland, S.; Mancini, S.J.; Salt, I.P. Role of AMP-activated protein kinase in adipose tissue metabolism and inflammation. Clin. Sci. 2013, 124, 491–507.
- Thors, B.; Halldórsson, H.; Thorgeirsson, G. eNOS activation mediated by AMPK after stimulation of endothelial cells with histamine or thrombin is dependent on LKB1. Biochim. Et Biophys. Acta 2011, 1813, 322–331.
- Meziat, C.; Boulghobra, D.; Strock, E.; Battault, S.; Bornard, I.; Walther, G.; Reboul, C. Exercise training restores eNOS activation in the perivascular adipose tissue of obese rats: Impact on vascular function. Nitric Oxide 2019, 86, 63–67.
- Aldiss, P.; Lewis, J.E.; Lupini, I.; Boocock, D.J.; Miles, A.K.; Ebling, F.J.; Budge, H.; Symonds, M.E. Exercise does not induce browning of WAT at thermoneutrality and induces an oxidative, myogenic signature in BAT. bioRxiv 2019, 649061.
- DeVallance, E.; Branyan, K.W.; Lemaster, K.C.; Anderson, R.; Marshall, K.L.; Olfert, I.M.; Smith, D.M.; Kelley, E.E.; Bryner, R.W.; Frisbee, J.C.; et al. Exercise training prevents the perivascular adipose tissue-induced aortic dysfunction with metabolic syndrome. Redox Biol. 2019, 26, 101285.
- Aghamohammadzadeh, R.; Greenstein, A.S.; Yadav, R.; Jeziorska, M.; Hama, S.; Soltani, F.; Pemberton, P.W.; Ammori, B.; Malik, R.A.; Soran, H.; et al. Effects of Bariatric Surgery on Human Small Artery Function: Evidence for Reduction in Perivascular Adipocyte Inflammation, and the Restoration of Normal Anticontractile Activity Despite Persistent Obesity. J. Am. Coll. Cardiol. 2013, 62, 128–135.