Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Lindsay Dong and Version 1 by Fernando Mendes.
Acute myeloid leukemia is an aggressive and very lethal blood tumor. It represents a substantial percentage of leukemia patients, as well as leukemic deaths. Immune checkpoint inhibition (ICI) has emerged as a therapeutic option for acute myeloid leukemia (AML) for patients that suffer from relapsed or high-risk disease, or patients ineligible for standard therapy.
- acute myeloid leukemia
- treatment
- immune checkpoint
1. Introduction
Acute myeloid leukemia (AML) is a bone marrow malignancy, characterized by the expansion and differentiation of myeloid progenitor cells [1,2][1][2]. AML represents around 90 percent of leukemia cases in adults and accounts for 62 percent of leukemic deaths [3,4,5][3][4][5]. This clonal hematopoietic stem cell disorder affects people of all ages; nevertheless, its incidence increases in older adults, reporting 68 years as the median age at diagnosis [6].
AML can result from an underlying hematological disorder, prior chemotherapy, or certain chemical exposures, or, as de novo malignancy [2[2][7],7], due to genetic alterations, through well-characterized chromosomal translocations or isolated molecular changes [7,8][7][8]. The criteria to diagnose AML are either the identification of more than 20% of myeloid blasts in the bone marrow or peripheral blood or the detection of specific cytogenetic abnormalities, as observed in Figure 1 [7][9].
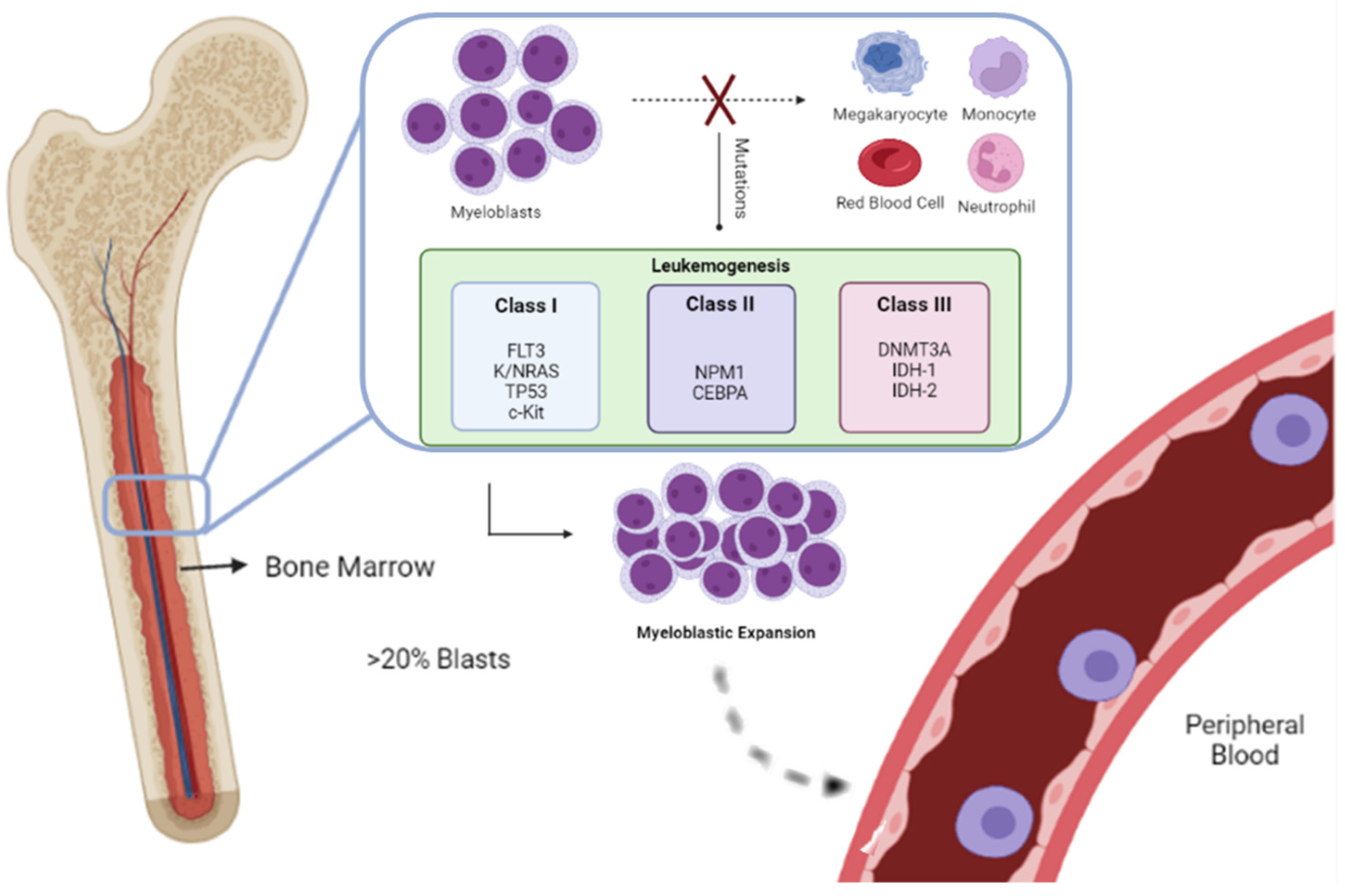
Figure 1. In Acute myeloid leukemia, due to genetic alterations in the blood cell precursors, there is an overproduction of neoplastic clonal myeloid cells, resulting in their accumulation in the bone marrow, namely, the FMS-like tyrosine kinase 3 (FLT3), Kirsten rat sarcoma viral oncogene homolog/neuroblastoma rat sarcoma viral oncogene homolog (K/NRAS), tumor protein 53 (TP53), tyrosine-protein kinase (c-KIT), nucleophosmin 1 (NPM1), CCAAT enhancer-binding protein alpha (CEBPA), DNA methyltransferase 3 alpha (DNMT3A), ten-eleven translocation-2 (TET2), isocitrate dehydrogenases 1 and 2 (IDH-1 and IDH-2). Myeloid blasts fail to differentiate into monocytes, megakaryocytes, neutrophils, and red blood cells. [7The expansion, over 20%, initially is in the bone marrow; however, in a later stage of the disease,9] blasts are detected in peripheral blood.
The subtypes of this pathology are classified by the World Health Organization. This classification system incorporates genetic information, morphology, immunophenotype, and clinical presentation. Distinguishing into six diverse groups: AML with recurrent genetic abnormalities, AML with three myelodysplasia-related changes, Therapy-related myeloid neoplasms, AML Not Otherwise Specified, Myeloid sarcoma, and Myeloid proliferations related to Down syndrome [10].
A two-hit model of leukemogenesis was developed to enable the classification of various mutations. Class I mutations, such as FMS-like tyrosine kinase 3 (FLT3), Kirsten rat sarcoma viral oncogene homolog/neuroblastoma rat sarcoma viral oncogene homolog (K/NRAS), tumor protein 53 (TP53), and c-KIT, result in the activation of pro-proliferative pathways. These must happen simultaneously to class II mutations, such as nucleophosmin 1 (NPM1) and CCAAT enhancer-binding protein alpha (CEBPA) to commit normal differentiation and develop leukemia. This model currently considers the alterations in the epigenetic regulation, the third class of mutations, in genes related to DNA methylation such as DNA methyltransferase 3 alpha (DNMT3A), ten-eleven translocation-2 (TET2), isocitrate dehydrogenases 1 and 2 (IDH-1 and IDH-2) [8,11][8][11].
The understanding of genetic abnormalities is key for both stratifying patients and, determining appropriate treatment. The standard treatment is chemotherapy, divided into induction therapy, and consolidation therapy. Both treatment and response vary according to the patient’s age, coexistence with other diseases, and genetic alterations [12,13,14][12][13][14].
Consolidation chemotherapy is administrated after intensive induction therapy to remove residual leukemic cells. After chemotherapy-induced remission, post-remission therapy must avoid relapse, a stem cell transplant in younger patients, or low-dose chemotherapy in older ones [14]. Patients younger than 60 years, present a 60% to 70% rate of complete remission (CR) with chemotherapy. Nevertheless, the cure rate is around 35% to 40%, which is considerably low. Regarding older patients, and patients who suffer from adverse cytogenetic risk, these present a lower CR rate, of 35% to 50%, therefore an even lower cure rate, of 10% or less. This lower response is due to either the inability to tolerate intensive chemotherapy or the higher rates of resistance to chemotherapy [20,21][15][16].
2. Immune System in Acute Myeloid Leukemia
Nowadays, the immune system has a significant role in tumor cell elimination. In AML, a common problem is tumor cell immune escape, being an effective survival mechanism [35,36][17][18]. AML blast cells have the capacity to block immune system communication and weaken T-cells, namely in their cytotoxic activity. The immune escape of AML blast cells in bone marrow can occur due to the burying of the malignant cells in the immune system or influencing negatively the different immune cells [37,38][19][20]. T-cells can be classified into subgroups, such as T-cell cytotoxic, T cell helper, and Regulatory T-cells (Tregs). This last one can reduce inflammatory reactions, by secreting anti-inflammatory cytokines as well as suppressing cytotoxic T-cells activity [35,39][17][21]. Therefore, Tregs’ may provide protection to AML cells from the immune system. AML blasts express programmed death ligand 1 (PD-L1) as an immune checkpoint inhibitor, and produce indolamine-2,3-dioxygenase (IDO) and reactive oxygen species, which induce differentiation towards Tregs, facilitating disease progression [39,40,41,42][21][22][23][24]. On the other hand, T-cell anergy can also be induced through T-cell immunoglobulin and mucin domain 3 (TIM3), which binds to and is activated by galectin-9, which is highly expressed in AML blasts [43][25]. Another suppressive molecule is an inducible T-cell co-stimulator ligand (ICOSL) and IDO’s that contribute to Tregs expansion and an immunosuppressive environment and at the same time restrict cytotoxic activity [36,44,45][18][26][27]. High expression levels of PD1/PD-L1 and TIM3 are correlated with a worse prognosis in AML [36,46][18][28]. Besides decreasing T-cell cytotoxic activity, T-cell exhaustion is also described as related to significant changes in the AML microenvironment metabolically, which is described as being rich in glutamine and poor in arginine [47,48][29][30]. Another white cell subset, the B cells have not been studied profoundly; although, a recent study showed that memory B-cells have a negative impact on survival, while increased numbers of naïve B-cells seem to have a positive impact [49][31]. AML blasts can modify the function of the innate immune system cells, namely, macrophages and Natural Killer (NK) cells, downregulating surface molecules required for NK cells recognition through receptor natural killer group 2 member D (NKG2D), and issue altered NKG2D-ligands, therefore contributing to the reduced cytotoxic activity of NK cells; one more mechanism to evade NK-cell recognition is the interferon α (IFN-α) reduces NKG2D by AML blasts, and in this way escaping destruction by nu NK cells [50,51,52,53][32][33][34][35]. Moreover, AML blasts can promote macrophages’ shift toward M2 polarization (immunosuppressive profile), associated with the promotion of tissue repair and angiogenesis [32,54][36][37]. Myeloid-derived suppressor cells (MDSC) are also influenced by AML blast cells, inducing T-cell inactivity through numerous mechanisms from PD-L1 expression to cytokines secretion such as interleukin (IL)-10 and/or tumor growth factor- β (TGF-β). Jointly with Tregs and M2-macrophages, MDSC has high numbers in AML patients [55][38], and, consequently, seems to be a risk factor for disease progression [56][39]. Ultimately, AML blasts seem to have a defective antigen-presentation, consequently downregulating the expression of human leukocyte antigens (HLA), assisting to make them unseen to the immune cells [57][40].3. Acute Myeloid Leukemia Therapy
3.1. Chemotherapy
Chemotherapy is still the standard therapy used for AML. Tumor cell death is caused not only by cytostatic effects but also by the restimulation of the immune surveillance with a direct immune response towards tumor cells [58,59][41][42]. The immune response against cancer cells is increased through various processes, from enhancing antigen uptake and chemotactic response via macrophages and dendritic cells to augmenting recognition to the immune system and increasing tumor susceptibility to immune-mediated cytotoxicity [58,59,60][41][42][43]. Provoking an immune response with chemotherapy is crucial to inducing immunogenic cell death (ICD) rather than a non-immunogenic cell death (non-ICD), also known as apoptosis. To achieve this, therapy should direct to a pre-apoptotic exposure at the cell surface of calreticulin (CRT), secretion of ATP during the blebbing phase of apoptosis, and the cell death-associated release of the non-histone chromatin protein high-mobility group box 1 (HMGB1) [61][44]. As AML patients present a spontaneous exposure of CRT in tumor cells, this predicts an antitumor T cell response and improves patient survival [58][41]. However, AML is a heterogeneous and aggressive disease that may show an initial response to chemotherapy, but if not completely eradicated becomes progressively more resistant and relapses [14,62][14][45]. To overcome this adaptive resistance and reduce the risk of relapse, the combination of immune checkpoint inhibitors with traditional chemotherapy is currently being experimented [58][41].3.2. Allogeneic Stem Cell Transplantation
Graft-Versus-Leukemia Effect and Graft-Versus-Host Disease
For patients with AML, the cure depends not only on the intensity of the conditioning treatment given before the transplantation but also on the immune-mediated graft-versus-leukemia (GvL) effect [63][46]. It is currently established that the donor immune system can mediate an effective GvL effect in many hematological malignancies [64,65][47][48]. In an allogeneic hematopoietic stem cell transplantation, the immune system of the donor aims to eliminate the residual leukemia cells that persist after prior (radio)chemotherapy. This immune-mediated response is acknowledged as graft-versus-leukemia, which represents a positive response in treating cancer. Donor T cells recognize cancer cells through the binding of T cell receptors to major histocompatibility (MHC) molecules present on the surface of cancer cells. MHC is known as human leukocyte antigens (HLA), so the donor is chosen based on the matching of HLA alleles, reducing graft rejection [63,66][46][49]. On the other hand, donor alloimmune response can target patients’ healthy tissues, a response known as graft-versus-host disease (GvHD). GvHD is a multi-system disorder that commonly initially affects the skin, then the gastrointestinal tract, liver, and lungs, and in a later stage, can affect almost any organ. GvHD and GvL, regularly happen, however, do not always occur simultaneously, suggesting that is possible to promote GvL without GvHD [63,65][46][48].4. Immune Checkpoint Blockade in Acute Myeloid Leukemia
Checkpoint inhibition is a breakthrough in the treatment of AML patients [76][50]. The evolution in the molecular understanding of AML and in tumor immunology allows the targeting of AML with immunotherapeutic strategies such as immune checkpoint inhibitors [66][49]. The clinical application of checkpoint blockers has achieved modest results both as a monotherapy and when combined with chemotherapy [73,74][51][52]. Relapsed hematological diseases and post-allo-HSCT have limited treatment options. CTLA-4 blockade with ipilimumab showed clinically noteworthy remissions in patients with recurrent cancer after transplantation. No objective responses were achieved at the 3 mg/kg dosage; however, 32% who received the 10 mg/kg had a response; this accomplishment suggests that the antibody dosage may be significant after transplantation. Five patients had a complete remission and four patients who had a response maintained a remission lasting over a year. This observation suggests that CTLA-4 blockade may be effective post-allo-HSCT. Immune-related adverse events associated with ipilimumab were observed in a few patients (six/total of the patients in the study). Once the study involved patients three or more months after transplantation, it was not possible to conclude the safety of ipilimumab in the early post-transplantation period. Outlining this study, CTLA-4 blockade was an advantageous approach for the treatment of these patients, presenting durable complete remissions, even in patients with refractory myeloid cancers [73][51]. For high-risk AML patients not eligible for allo-HSCT, nivolumab treatment presented a recurrence-free survival similar to the mean values observed in the literature. However, a promising overall survival was observed in these patients. Furthermore, it also showed a significant effect in eradicating MRD and extending remissions, as a single agent. Nevertheless, immune-related adverse events occurred recurrently. In this setting, the provided data do not support the use of single-agent nivolumab; although, it offers background and viability for the incorporation of an immune checkpoint blockade in combination trials, as maintenance therapy for high-risk AML patients. The most adequate post-remission therapy in high-risk AML continues to be allo-HSCT [75][53]. The first nivolumab prospective trial for post-allo-HSCT for relapsed hematological malignancies identified the maximum tolerated dose (MTD) as a low dose of 0.5 mg/kg. PD-1 blockade involves a risk of inducing immune-related adverse events and GvHD, as observed in 39% of the patients in this study, with two fatal patients, despite the low doses of nivolumab [76][50]. Not only the induced immune-related adverse events but also the demand for managing these toxicities is similar to other retrospective studies performed previously [82,83][54][55]. The combination of nivolumab with idarubicin plus high dose cytarabine, both chemotherapeutic agents, as the frontline therapy in AML patients and high-risk myelodysplastic syndromes (MDS), led to objective responses in 80% of the patients, 64% of which CR. It was hypothesized that the induction of an antileukemic immune response through nivolumab would be efficient in increasing the durability of the response, or even that nivolumab could eliminate minimal residual disease.5. Conclusions
AML treatment with monotherapy, with Ipilimumab, in relapsed hematologic malignancies, after allo-HSCT, is a worthy therapeutic approach due to the high percentage of complete remissions observed (42%). The treatment of patients in the same clinical circumstances, with nivolumab, even at a low dosage, showed significant immune-mediated toxicities in myeloid malignancies. Further investigation of this checkpoint blocker, in this setting could evaluate better strategies to decrease toxicity and increase clinical benefit. The use of nivolumab, as single-agent maintenance therapy in high-risk AML presented an interesting overall survival in these patients; however, the recurrence-free survival duration was shorter than the described results in the literature. These results, do not encourage the use of nivolumab as a single agent, in this setting; nevertheless, these data suggest that the use of checkpoint blockers in combination therapy may provide a better outcome. In combination therapy, with chemotherapeutic agents, the use of nivolumab in newly diagnosed AML or high-risk MDS is feasible and safe in younger patients with AML and 80% of the patients presented objective responses. Meanwhile, the approach of AML and MDS patients with checkpoint blockage before allo-HSCT and the use of a GvHD prophylaxis has shown improvement in the transplantation outcomes, verifying a comparable difference between the use of PCTy and no-PCTy as prophylaxis.References
- Gómez-Llobell, M.; Peleteiro Raíndo, A.; Climent Medina, J.; Gómez Centurión, I.; Mosquera Orgueira, A. Immune Checkpoint Inhibitors in Acute Myeloid Leukemia: A Meta-Analysis. Front. Oncol. 2022, 12, 882531.
- Pelcovits, A.; Niroula, R. Acute Myeloid Leukemia: A Review. Rhode Isl. Med. J. 2020, 103, 38–40.
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953.
- Almeida, A.L.S.; de Azevedo, I.C.; Pinto, D.P.D.S.R.; Vitor, A.F.; Euzébia, V.; Júnior, M.A.F. Clinical and epidemiological aspects of leukemias Aspectos clínicos y epidemiológicos de las leucemias. Rev. Cuba. Hematol. Inmunol. Hemoter. 2017, 33, 1–14.
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Zeidan, A.M. Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev. 2019, 36, 70–87.
- Carter, J.L.; Hege, K.; Yang, J.; Kalpage, H.A.; Su, Y.; Edwards, H.; Hüttemann, M.; Taub, J.W.; Ge, Y. Targeting multiple signaling pathways: The new approach to acute myeloid leukemia therapy. Signal Transduct. Target. Ther. 2020, 5, 288.
- Strickland, S.A.; Vey, N. Diagnosis and treatment of therapy-related acute myeloid leukemia. Crit. Rev. Oncol. Hematol. 2022, 171, 103607.
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441.
- Narayanan, D.; Weinberg, O.K. How I investigate acute myeloid leukemia. Int. J. Lab. Hematol. 2020, 42, 3–15.
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood Am. Soc. Hematol. 2016, 127, 2391–2405.
- Newell, L.F.; Cook, R.J. Advances in acute myeloid leukemia. BMJ 2021, 375, n2026.
- Acheampong, D.O.; Adokoh, C.K.; Asante, D.B.; Asiamah, E.A.; Barnie, P.A.; Bonsu, D.O.; Kyei, F. Immunotherapy for acute myeloid leukemia (AML): A potent alternative therapy. Biomed. Pharmacother. 2018, 97, 225–232.
- Przespolewski, A.; Szeles, A.; Wang, E.S. Advances in immunotherapy for acute myeloid leukemia. Futur. Oncol. 2018, 14, 963–978.
- Beyar-Katz, O.; Gill, S. Novel approaches to acute myeloid leukemia immunotherapy. Clin. Cancer Res. 2018, 24, 5502–5515.
- Almeida, A.M.; Ramos, F. Acute myeloid leukemia in the older adults. Leuk. Res. Rep. 2016, 6, 1–7.
- Albring, J.C.; Inselmann, S.; Sauer, T.; Schliemann, C.; Altvater, B.; Kailayangiri, S.; Rössig, C.; Hartmann, W.; Knorrenschild, J.R.; Sohlbach, K.; et al. PD-1 checkpoint blockade in patients with relapsed AML after allogeneic stem cell transplantation. Bone Marrow Transplant. 2017, 52, 317–320.
- Mendes, F.; Domingues, C.; Rodrigues-Santos, P.; Abrantes, A.M.; Goncalves, A.C.; Estrela, J.; Encarnacao, J.; Pires, A.S.; Laranjo, M.; Alves, V. The role of immune system exhaustion on cancer cell escape and anti-tumor immune induction after irradiation. Biochim. Biophys. Acta-Rev. Cancer 2016, 1865, 168–175.
- Menter, T.; Tzankov, A. Tumor Microenvironment in Acute Myeloid Leukemia: Adjusting Niches. Front. Immunol. 2022, 13, 34–43.
- Lim, S.H.; Worman, C.P.; Jewell, A.P.; Goldstone, A.H. Cellular cytotoxic function and potential in acute myelogenous leukaemia. Leuk. Res. 1991, 15, 641–644.
- Le Dieu, R.; Taussig, D.C.; Ramsay, A.G.; Mitter, R.; Miraki-Moud, F.; Fatah, R.; Lee, A.M.; Lister, T.A.; Gribben, J.G. Peripheral blood T cells in acute myeloid leukemia (AML) patients at diagnosis have abnormal phenotype and genotype and form defective immune synapses with AML blasts. Blood 2009, 114, 3909–3916.
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular mechanisms of treg-mediated T cell suppression. Front. Immunol. 2012, 3, 51.
- Mansour, I.; Zayed, R.A.; Said, F.; Latif, L.A. Indoleamine 2,3-dioxygenase and regulatory T cells in acute myeloid leukemia. Hematology 2016, 21, 447–453.
- Robinson, A.J.; Davies, S.; Darley, R.L.; Tonks, A. Reactive Oxygen Species Rewires Metabolic Activity in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 458.
- Zhang, L.; Gajewski, T.F.; Kline, J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood 2009, 114, 1545–1552.
- Rezaei, M.; Tan, J.; Zeng, C.; Li, Y.; Ganjalikhani-Hakemi, M. TIM-3 in Leukemia; Immune Response and Beyond. Front. Oncol. 2021, 11, 3939.
- Arandi, N.; Ramzi, M.; Safaei, F.; Monabati, A. Overexpression of indoleamine 2,3-dioxygenase correlates with regulatory T cell phenotype in acute myeloid leukemia patients with normal karyotype. Blood Res. 2018, 53, 294.
- Han, Y.; Dong, Y.; Yang, Q.; Xu, W.; Jiang, S.; Yu, Z.; Yu, K.; Zhang, S. Acute Myeloid Leukemia Cells Express ICOS Ligand to Promote the Expansion of Regulatory T Cells. Front. Immunol. 2018, 9, 2227.
- Tettamanti, S.; Pievani, A.; Biondi, A.; Dotti, G.; Serafini, M. Catch me if you can: How AML and its niche escape immunotherapy. Leukemia 2022, 36, 13–22.
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758.
- Nabe, S.; Yamada, T.; Suzuki, J.; Toriyama, K.; Yasuoka, T.; Kuwahara, M.; Shiraishi, A.; Takenaka, K.; Yasukawa, M.; Yamashita, M. Reinforce the antitumor activity of CD8+ T cells via glutamine restriction. Cancer Sci. 2018, 109, 3737–3750.
- Cheng, Y.; Wang, X.; Qi, P.; Liu, C.; Wang, S.; Wan, Q.; Liu, Y.; Su, Y.; Jin, L.; Liu, Y.; et al. Tumor Microenvironmental Competitive Endogenous RNA Network and Immune Cells Act as Robust Prognostic Predictor of Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 919.
- Paczulla, A.M.; Rothfelder, K.; Raffel, S.; Konantz, M.; Steinbacher, J.; Wang, H.; Tandler, C.; Mbarga, M.; Schaefer, T.; Falcone, M.; et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 2019, 572, 254–259.
- Baragaño Raneros, A.; Martin-Palanco, V.; Fernandez, A.F.; Rodriguez, R.M.; Fraga, M.F.; Lopez-Larrea, C.; Suarez-Alvarez, B. Methylation of NKG2D ligands contributes to immune system evasion in acute myeloid leukemia. Genes Immun. 2015, 16, 71–82.
- Zhu, L.; Kong, Y.; Zhang, J.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; Palmisiano, N.D.; Wang, M.; Jia, B.; Bayerl, M.; et al. Blimp-1 impairs T cell function via upregulation of TIGIT and PD-1 in patients with acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 124.
- Kong, Y.; Zhu, L.; Schell, T.D.; Zhang, J.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; George, M.R.; Zeng, H.; Zheng, H.; et al. T-cell immunoglobulin and ITIM domain (TIGIT) associates with CD8+ T-cell exhaustion and poor clinical outcome in AML patients. Clin. Cancer Res. 2016, 22, 3057–3066.
- Al-Matary, Y.S.; Botezatu, L.; Opalka, B.; Hönes, J.M.; Lams, R.F.; Thivakaran, A.; Schütte, J.; Köster, R.; Lennartz, K.; Schroeder, T.; et al. Acute myeloid leukemia cells polarize macrophages towards a leukemia supporting state in a growth factor independence 1 dependent manner. Haematologica 2016, 101, 1216–1227.
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555.
- Yang, Y.; Li, C.; Liu, T.; Dai, X.; Bazhin, A.V. Myeloid-Derived Suppressor Cells in Tumors: From Mechanisms to Antigen Specificity and Microenvironmental Regulation. Front. Immunol. 2020, 11, 1371.
- Kittang, A.O.; Kordasti, S.; Sand, K.E.; Costantini, B.; Kramer, A.M.; Perezabellan, P.; Seidl, T.; Rye, K.P.; Hagen, K.M.; Kulasekararaj, A.; et al. Expansion of myeloid derived suppressor cells correlates with number of T regulatory cells and disease progression in myelodysplastic syndrome. Oncoimmunology 2016, 5, e1062208.
- Christopher, M.J.; Petti, A.A.; Rettig, M.P.; Miller, C.A.; Chendamarai, E.; Duncavage, E.J.; Klco, J.M.; Helton, N.M.; O’Laughlin, M.; Fronick, C.C.; et al. Immune Escape of Relapsed AML Cells after Allogeneic Transplantation. N. Engl. J. Med. 2018, 379, 2330–2341.
- Stahl, M.; Goldberg, A.D. Immune Checkpoint Inhibitors in Acute Myeloid Leukemia: Novel Combinations and Therapeutic Targets. Curr. Oncol. Rep. 2019, 21, 37.
- Valent, P.; Bauer, K.; Sadovnik, I.; Smiljkovic, D.; Ivanov, D.; Herrmann, H.; Filik, Y.; Eisenwort, G.; Sperr, W.R.; Rabitsch, W. Cell-based and antibody-mediated immunotherapies directed against leukemic stem cells in acute myeloid leukemia: Perspectives and open issues. Stem Cells Transl. Med. 2020, 9, 1331–1343.
- Thol, F.; Ganser, A. Treatment of Relapsed Acute Myeloid Leukemia. Curr. Treat. Options Oncol. 2020, 21, 66.
- Pitt, J.M.; Kroemer, G.; Zitvogel, L. Immunogenic and non-immunogenic cell death in the tumor microenvironment. Adv. Exp. Med. Biol. 2017, 1036, 65–79.
- Wu, J.; Waxman, D.J. Immunogenic chemotherapy: Dose and schedule dependence and combination with immunotherapy. Cancer Lett. 2018, 419, 210–221.
- Baron, F.; Labopin, M.; Savani, B.N.; Beohou, E.; Niederwieser, D.; Eder, M.; Potter, V.; Kroeger, N.; Beelen, D.; Socie, G.; et al. Graft-versus-host disease and graft-versus-leukaemia effects in secondary acute myeloid leukaemia: A retrospective, multicentre registry analysis from the Acute Leukaemia Working Party of the EBMT. Br. J. Haematol. 2020, 188, 428–437.
- Loke, J.; Malladi, R.; Moss, P.; Craddock, C. The role of allogeneic stem cell transplantation in the management of acute myeloid leukaemia: A triumph of hope and experience. Br. J. Haematol. 2020, 188, 129–146.
- Haverkos, B.M.; Abbott, D.; Hamadani, M.; Armand, P.; Flowers, M.E.; Merryman, R.; Kamdar, M.; Kanate, A.S.; Saad, A.; Mehta, A.; et al. PD-1 blockade for relapsed lymphoma post-allogeneic hematopoietic cell transplant: High response rate but frequent GVHD. Blood 2017, 130, 221–228.
- Sweeney, C.; Vyas, P. The Graft-Versus-Leukemia Effect in AML. Front. Oncol. 2019, 9, 1217.
- Davids, M.S.; Kim, H.T.; Costello, C.; Herrera, A.F.; Locke, F.L.; Maegawa, R.O.; Savell, A.; Mazzeo, M.; Anderson, A.; Boardman, A.P.; et al. A multicenter phase 1 study of nivolumab for relapsed hematologic malignancies after allogeneic transplantation. Blood 2020, 135, 2182–2191.
- Davids, M.S.; Kim, H.T.; Bachireddy, P.; Costello, C.; Liguori, R.; Savell, A.; Lukez, A.P.; Avigan, D.; Chen, Y.B.; McSweeney, P.; et al. Ipilimumab for Patients with Relapse after Allogeneic Transplantation. N. Engl. J. Med. 2016, 375, 143–153.
- Vago, L.; Gojo, I. Immune escape and immunotherapy of acute myeloid leukemia. J. Clin. Investig. 2020, 130, 1552–1564.
- Reville, P.K.; Kantarjian, H.M.; Ravandi, F.; Jabbour, E.; DiNardo, C.D.; Daver, N.; Pemmaraju, N.; Ohanian, M.; Alvarado, Y.; Xiao, L.; et al. Nivolumab maintenance in high-risk acute myeloid leukemia patients: A single-arm, open-label, phase II study. Blood Cancer J. 2021, 11, 60.
- Isidori, A.; Cerchione, C.; Daver, N.; DiNardo, C.; Garcia-Manero, G.; Konopleva, M.; Jabbour, E.; Ravandi, F.; Kadia, T.; Burguera, A.D.L.F.; et al. Immunotherapy in Acute Myeloid Leukemia: Where We Stand. Front. Oncol. 2021, 11, 656218.
- Bewersdorf, J.P.; Stahl, M.; Zeidan, A.M. Immune checkpoint-based therapy in myeloid malignancies: A promise yet to be fulfilled. Expert Rev. Anticancer Ther. 2019, 19, 393–404.
More