Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Trang Truong and Version 4 by Catherine Yang.
Drug repurposing offers a more efficient pathway compared with de novo drug discovery with lower cost and less risk. Various computational approaches have been applied to mine the vast amount of biomedical data generated over recent decades. Aligned well with the poly-pharmacology paradigm shift in drug discovery, network-based approaches offer great opportunities to discover repurposing candidates for complex psychiatric disorders.
- drug repurposing
- psychiatric disorders
- medications
1. Introduction
Network-based interpretation comprises three major steps from understanding to predicting and possible manipulating biological systems: (1) network inference (reconstruction of network relationships from biomedical data, mostly from high-throughput assays), (2) network analysis (harnessing the topological relationships of networks), (3) network modelling (dynamic representations of time-course perturbations of network elements under different conditions) [1][2]. Most studies so far have utilised the first two steps for static networks, but very few have advanced to dynamic network modelling [3].
A network inference approach involves “simplifying” complex systems by describing them as a map of nodes connected by edges denoting their relationships or interactions [4] (Figure 12). While networks can represent a wide range of biological processes, in the context of drug discovery research, nodes are generally molecular targets (genes, proteins), compounds (drugs) or diseases, with their relationships inferred from structural interactions (e.g., protein–protein interactions), correlation (e.g., co-expression networks) or conditional dependences (e.g., Bayesian networks) [5]. Many real-world networks including biological networks, tend to exhibit scale-free properties, which means only a minority of nodes have a greater number of neighbours than average (“hubs”), while most nodes only have a few connections [6][7][8]. Selective targeting of hubs can therefore cause much greater impact on the function of the networks than those modulations on peripheral nodes, making hubs ideal drug targets [9].
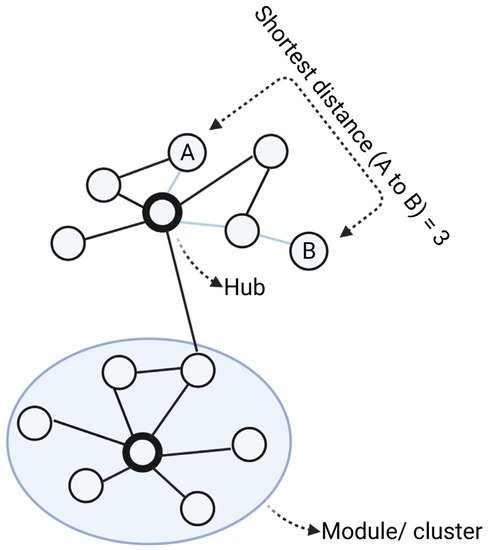
Figure 12. Main elements of a network. In the network, nodes (circles) are connected via edges (lines). For biological networks, nodes are usually biological entities (genes, proteins) and edges denote their relationships (interaction, association, similarity). From the networks, modules are clusters of closely connected nodes. Degree is the number of direct connections a node has to other nodes. Hubs are nodes with the highest degrees in the networks, meaning they have the highest number of connections. The shortest distance between node A and B is the path with the minimum number of edges from A to B. Created with BioRender.com (accessed on 2 June 2022).
Network-based drug repurposing efforts are generally based on Swanson’s ABC model to retrieve unknown latent knowledge from multiple sources of data incorporated in the networks [10]. An assumption of this approach is that when term A is connected to term B, and term B is connected to term C, it can be assumed that terms A and C are also connected. For example, an indirect link between drug and disease can be inferred from a direct drug-target connection and a direct target-disease connection. In the ABC model, A and C must originate from different domains to yield new knowledge, and B can include multiple steps to abridge from A to C (A → B1 → B2 … Bn → C) [11][12] (Figure 23).
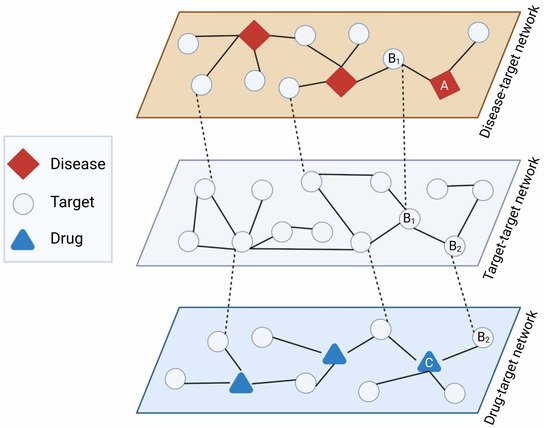
Figure 23. ABC model for network-based drug repurposing. Latent repurposing relationships can be inferred from multiple layers of network-based knowledge such as disease-target (diseasome), target–target (e.g., protein interactome), and drug–target interactions. As an example, disease A has target B1 exhibiting direct interaction with target B2 which in turn is targeted by drug C, suggesting drug C might be relevant for disease A (A → B1 → B2 → C). Created with BioRender.com (accessed on 2 June 2022).
Another common approach is “guilt-by-association” (GBA), which uses similarity measures to suggest new disease indications for drugs [13]. There are two main assumptions of GBA: (1) if two diseases share a significant number of characteristics (e.g., indications, medical descriptions, mechanisms), a drug known to treat one of them may also treat the other (Figure 34A); and (2) if a drug with unknown indications and another drug with known indications share similar properties (e.g., chemical structures, transcriptional effects), they may have the same indication profile (Figure 34B). The major challenge of this approach would be how to define the robust similarity metric between drugs or diseases that concurs with similarity in mechanisms of action.
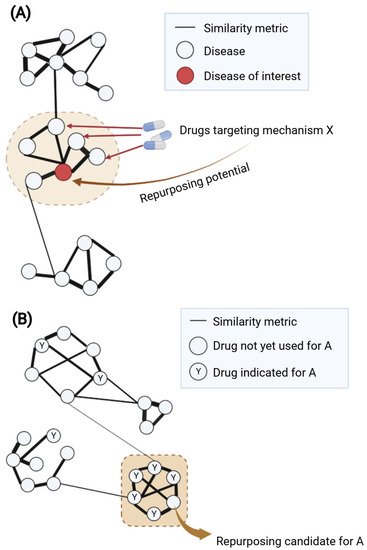
Figure 34. Guilt-by-association for network-based drug repurposing using (A) disease–disease or (B) drug–drug similarity. (A) Disease–disease similarity is generally inferred from one or several disease-related properties such as overlapping disease genes, symptoms or comorbidities. A weighted disease network (diseasome) can be built based on the similarity metric; herein, modules of similar nodes (diseases) can be identified. The module containing the disease of interest (highlighted in the brown dashed circle) might suggest potential shared mechanism(s) for repurposing drugs. Within this module, if multiple connected diseases have known drugs with similar mechanism X, such drugs might be repurposed for the disease of interest. (B) Drug–drug similarity can be calculated based on one or several properties such as chemical structures, targets, side effects or transcriptional profiles. Using the similarity metric as the weight of edges for network construction, ones can identify modules of highly similar nodes (drugs) suggesting similar mechanisms of action. When considering in the context of a certain disease A, it would be of interest to focus on the module containing multiple known drugs for disease A (highlighted as brown dashed square). Within such a module, a drug that has yet to be used for disease A might be a potential repurposing candidate due to its high similarity with other drugs used for disease A. Created with BioRender.com (accessed on 2 June 2022).
Data for network construction can be sourced from experimental data (e.g., high throughput screening), text mining or databases (e.g., phenotypic profiles, protein interactions). Text mining is also the main strategy of literature-based drug repurposing, which shares many integrative opportunities with network-centric approaches. Hence, readers can refer to previous reviews in this domain for an in-depth methodological presentation [14][15]. The advantage of network-based approaches is the possible integration of multiple data layers to complement the incompleteness of each domain’s knowledge. Therefore, studies using network-based drug repurposing tend to utilise multiple data sources rather than one. There are various ways of data incorporation to find repurposing insights as shown in Figure 45. However, one should consider the relevance to the disease of interest (e.g., data yielded from brain tissue versus muscle tissue) and the robustness of the evidence supporting such a relationship (e.g., experimental evidence versus co-expression). Multi-omics integration has been playing a major role in the current biological interpretation and readers can refer to previous reviews of specific updates and recommendations for this approach [16].
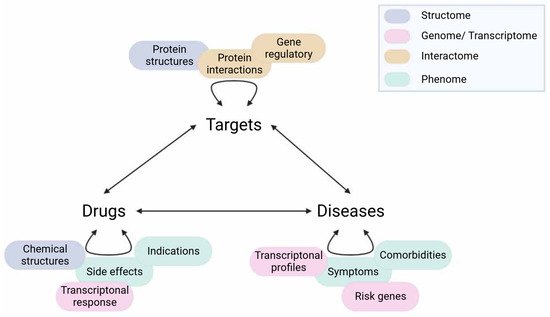
Figure 45. Different data sources for network-based drug repurposing. Curved arrows represent the associations of entities within one type (e.g., drug–drug). Multiple data sources (coloured correspondingly to their main domains such as transcriptome) can be applied to infer these associations, usually for the creation of similarity or interacting networks. Straight arrows represent the relationships between entities of different types (e.g., drug–target). For drug repurposing, the aim generally is to find a latent drug–disease connection, which can be achieved by taking the inference route from Drugs–Targets–Diseases (and vice- versa) as in the ABC model, or via Diseases–Diseases–Drugs (or Drugs–Drugs–Diseases) as in the GBA model. Created with BioRender.com (accessed on 2 June 2022).
2. Structural Data (Structome)
Structural data from compounds and biological entities such as proteins and RNAs have been extensively utilized in structure-based drug repurposing [17]. The conventional structure-based approach usually requires a few predefined specific target molecules, which is not suitable for psychiatric disorders with complex pathology as mentioned in Section 3. However, network-centric approaches can incorporate the structome as a layer of information in a non-biased way to find new indications for drugs. Tan et al. used descriptions of 3D chemical structures from PubChem to calculate the similarity profiles of 965 drugs [18]. The Tanimoto-based 3D similarity scores were then combined with gene semantic similarity information and drug–target interactions to construct a drug similarity network. From this GBA approach, Tan et al. predicted new indications for 143 drugs and missing indications for 42 drugs without Anatomical Therapeutic Chemical (ATC) codes (indications not yet listed in ATC database). Psychotropic drugs suggested for repurposing from this study included raloxifene (from postmenopausal osteoporosis to schizophrenia) and cyclobenzaprine (from muscle spasms to sleep disorders) [18]. Raloxifene has passed a phase 4 clinical trial in participants with schizophrenia [19][20] while a phase 2 clinical trial of cyclobenzaprine was terminated prematurely due to inadequate recruitment [21].
3. Genome
Using the phenotype-to-genotype concept, multiple large-scale genome-wide association studies (GWAS) have identified thousands of genetic variants across the genome associated with psychiatric disorders [22][23]. Disease-associated genes located in risk loci can be inferred from GWAS data and are usually used in network analysis as a filtering layer to prioritise targets relevant to the disease. Ganapathiraju et al. used schizophrenia-associated genes in combination with protein–protein interactions to create a schizophrenia interactome [24]. Such a disease-specific network can be harnessed for target identification and testing of repurposed agents [25]. However, a major limitation of using GWAS data is the lack of directionality, making it difficult to determine whether a risk gene is up- or down-regulated in the disease phenotype. Gaspar et al. partially addressed this shortcoming via the incorporation of the GWAS summary statistics with gene expression to predict expression levels in different tissues, which were incorporated with drug–target interactions to build a bipartite tissue-specific drug–target network for major depression [26] (Table 2).
4. Transcriptome
Among the wealth of “omics” data, transcriptomic profiling has emerged as an efficient source for computational drug repurposing due to its standardized data format, multiple comprehensive public databases, and possible implementation with network biology approaches for complex diseases [27][28][29]. The expression patterns of gene products that are connected by signalling cascades or protein complexes are expected to be more similar than those of random gene products [4][30]. With this premise, co-expression networks built upon multi-dimensional data such as transcriptomics have aided in the identification of latent mechanistic patterns of psychiatric disorders and their medications, which could be missed by conventional differential expression analysis [30][31].
Psychiatric disease-related transcriptional profiles, generally from post-mortem brain samples, can be readily obtained from experiments, public databases, or psychiatric-centric consortiums such as PsychENCODE and CommonMind [32][33]. The transcriptomic data can be used on its own (gene expression levels) or incorporated with GWAS data to predict genetically regulated gene expression. As an example of the former, Cabrera-Mendoza et al. used transcriptional profiles from post-mortem brain samples of substance-use disorder individuals with and without suicidal behaviour to build gene co-expression networks associated with each phenotype (Table 2). The hub genes from these networks were then subjected to drug–gene interaction testing using the DGIdb database [34] to identify drug repurposing candidates [35]. Integration of transcriptomic profiles with GWAS data was adopted by Rodriguez-López et al. for finding druggable targets in schizophrenia. The researcheuthors estimated polygenic scores based on predicted expression and associated these scores with co-expression modules to find relevant hub target genes for early intervention [36]. Gaspar et al. also applied the genetically predicted gene expression approach [26].
Major sources of drug-induced transcriptional profiles are generated from cell lines after treatment exposure, utilising seminal reference databases for drug responses such as Connectivity Map (CMap) [37] and the Library of Integrated Network-based Cellular Signatures (LINCS) [38]. While transcriptional profiles have been used extensively in signature-based drug repurposing for the generation and comparison of selective genes representing the phenotype of interest [28][39], their network-centric drug repurposing application is still very limited in psychiatry. An emerging systems-level approach constructing gene-regulatory networks associated with each drug treatment-cell line pair using CMap expression data can offer a comprehensive characterisation of the mechanism of action of drugs. Such a systems-level approach includes information on complex interactions between multiple entities, beyond the reductionist consideration of several signature genes [40][41].
The major challenge of using drug-induced gene expression in psychiatry is the lack of biological and pathological representation of the treated model systems. Transcriptional perturbations are highly context-dependent; hence, the cancerous cells used commonly in CMap and LINCS might not recapitulate the tissue-specific effects in neuronal or glial cells. The advancement in stem cell technology has propelled the generation of patient-derived induced pluripotent stem cells (iPSC), leading to the genesis of the NeuroLINCS center of omics data generation for human iPSC response in neurological diseases [42]. Since iPSCs carry the genetic information of the patients, they recapitulate the disease-related mutations that would be more representative for diseases with significant genetic factors such as psychiatric disorders [43].
5. Interactome
Interactomes encompass the functional interactions of biological components, which might include physical contact between proteins (protein–protein interaction networks), metabolites (metabolic networks), transcription factors and putative regulatory elements (gene regulatory networks) or functional relationships only such as phenotypic profiling networks (phenome networks) [4]. The interactome might be placed in specific biological contexts such as signalling pathways or disease-related pathways [44]. The functional interactome based on phenotypic profiles have been broadly applied for drug discovery and will be discussed separately in the context of phenome-based networks. Interactome networks tend to possess small world property: nodes are well connected with only a few paths required for the shortest distance (Figure 12). This holds highly relevant for functionally associated nodes, ensuring a quick flow of regulatory information passing between them [45]. With the premise that risk genes tend to be more connected in the network than a set of random genes, Kauppi et al. utilised the protein interactome to map drug targets of antipsychotic drugs with networks of schizophrenia risk genes (Table 2). Using network topological analysis of shortest distance, they found risk genes were significantly localised into a distinct module and overlapped with antipsychotic drug targets. Kauppi et al. then evaluated druggable risk genes without direct links to known antipsychotic drug targets to find potential novel targets for schizophrenia such as nicotinic acetylcholine receptor genes [46].
Given the key contribution of transcription factors in the modulation of gene expression and driving phenotypic perturbations, the transcriptional regulome has been employed by De Bastiani et al. for drug repurposing in bipolar disorders [47]. Their study inferred transcription factors–targets interactions via a reverse-engineering prediction algorithm applied on human prefrontal cortex microarray data. The transcription factor-centric network comprised of modules of gene targeted by each transcription factor, called “regulons”. Based on case-control transcriptomics data, gene set enrichment analysis (GSEA) was applied on the regulons to find enriched regulons in bipolar disorder. These regulons were used as gene expression signatures to query connectivity map for potential drug candidates reverting disease-related regulon signatures. Several compounds with known clinical relevance in bipolar disorders were identified such as antipsychotics (chlorpromazine, haloperidol) and antidepressants (maprotiline, mianserin, and desipramine). The enstrudy also found novel repurposing candidates including non-steroidal anti-inflammatory agents (meclofenamic acid, ketorolac, acetylsalicylsalicylic acid and diflorasone) and an antioxidant agent (trolox C)[47].
6. Phenome
The collection of phenotypic data collected from drug-induced (indications, side-effects) or disease-associated phenotypes (symptoms, disease genes) has been extensively used for drug repurposing with the availability of comprehensive public sources such as DrugBank and PharmGKB [48][49]. Zhou et al. built a drug side effect–gene system comprising two networks: drug phenotypic network of side effect profiles from SIDER [50] and protein interactome network from STRING [51]. The two networks were interconnected via drug-target associations from DrugBank [48]. Zhou et al. then applied this phenome-driven drug discovery system in finding repurposing agents for opioid use disorders. Rather than finding drugs targeting the pathological mechanism of the disorder, which is still mainly unknown, the system explored repurposing candidates sharing similar side effects or common targets with drugs causing or indicated for opioid use disorders. Using a network-based iterative algorithm, top-ranked repurposing candidates including tramadol, olanzapine, mirtazapine, bupropion and atomoxetine were identified with supporting clinical corroboration [52].
Psychiatric disorders tend to share mechanisms, such as pleiotropic genes associated with multiple disorders. By incorporating disease phenome and disease genome networks together, one can explore the common pathophysiology between diseases and infer potential reusable targets of one disease in a different disease. Such a disease-gene network was first proposed by Goh et al. as a “diseasome”—a bipartite graph including all known genetic disorders and disease genes connected by the association of genetic mutations to disorders [53]. Such a network can be interpreted for gene-gene similarity (connected if two genes share a disorder), or disease–disease similarity (linked if two disorders share a gene). While the specific application of diseasome in psychiatric disorders is still limited, Lüscher Dias et al. built a diseasome network considering multiple psychiatric and neurological disorders using text mining. They found several clusters shared by multiple disorders and their enriched functional annotations, e.g., depression with anxiety disorder (enriched for inflammatory response), bipolar disorder with schizophrenia (enriched for long-term potentiation and circadian entrainment). However, Lüscher Dias et al. did not consider common genes for their drug repurposing steps but focused on unique genes associated with each disorder as potential targets for the corresponding disorder (ABC model), shifting back to a single-disease context [54]. To theour knowledge, there have been no cases using disease–disease similarity networks for drug repurposing in psychiatric disorders. An example outside of psychiatry from Langhauser et al. demonstrated how the repurposing hypothesis can be generated from a disease–disease similarity network of the diseasome, even from seemingly distinct diseases [55]. They built diseasome networks for 132 diseases based on four different relationships: shared genes, protein interactome, common symptoms and co-morbidity. From the diseasome, Langhauser et al. found the cGMP signalling pathway was associated with a cluster of disease phenotypes including neurological, cardiovascular, metabolic and respiratory diseases. This GBA approach suggested cGMP modulators as treatments for diseases belonging to this cluster. Based on this premise, the researcheuthors repurposed soluble guanylate cyclase (sGC) activators—cGMP generation facilitators—from their exclusive indications for cardiovascular diseases to neurological disorders and successfully validated their neuroprotection effects in vivo [55].
7. Network-Based Drug Repurposing Platforms
There are various approaches to yield network-based repurposing insights from biomedical data if one would like to build networks from the ground up, which has been comprehensively reviewed [3][5][56]. However, there are several platforms that can serve as a “one-stop shop” for network repurposing with the incorporation of multiple biological datasets, pre-constructed networks, pre-set analyses for easy access and queries of existing or user-generated data: for example, GRAND, a web-based database of gene regulatory networks specific for disease- or drug-related phenotypes inferred from prior experimental data such as protein–protein interactions, transcriptional profiles, transcriptional factor binding motifs and miRNAs predicted targets [40]. Using similarity scores based on properties of inferred regulatory networks, the CLUEreg tool of GRAND allows users to query a list of “high-targeted” and “low-targeted” genes or transcriptional factors of the disease to identify single or combinations of compounds that might “reverse” aberrant regulatory patterns [40]. Other examples of open-sourced platforms include PharmOmics and NeDRex; the former is a knowledgebase supporting gene-network-based drug repurposing and the latter allows heterogeneous network construction to mine disease modules for drug prioritization [57][58]. While these platforms would be easy to use with curated networks, users are limited by the scope of the current platforms, and how regularly they are updated. Reproducibility would be a challenge especially with commercial platforms such as IBM Watson for Drug Discovery where detailed analysing workflows are not publicly accessible [59]. Moreover, most datasets incorporated were yielded from different domains such as oncology, weakening the robustness of interpretations in psychiatry.
References
- Albert, R.K. Network Inference, Analysis, and Modeling in Systems Biology. Plant Cell 2007, 19, 3327–3338.
- Kitano, H. Systems Biology: A Brief Overview. Science 2002, 295, 1662–1664.
- Recanatini, M.; Cabrelle, C. Drug Research Meets Network Science: Where Are We? J. Med. Chem. 2020, 63, 8653–8666.
- Vidal, M.; Cusick, M.E.; Barabási, A.-L. Interactome Networks and Human Disease. Cell 2011, 144, 986–998.
- Network Medicine: Complex Systems in Human Disease and Therapeutics; Harvard University Press: Cambridge, MA, USA, 2017.
- Barabási, A.-L.; Albert, R. Emergence of Scaling in Random Networks. Science 1999, 286, 509–512.
- Seebacher, J.; Gavin, A.C. SnapShot: Protein-protein interaction networks. Cell 2011, 144, 1000.
- Barabási, A.-L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113.
- Penrod, N.M.; Cowper-Sal-lari, R.; Moore, J.H. Systems genetics for drug target discovery. Trends Pharmacol. Sci. 2011, 32, 623–630.
- Swanson, D.R. Fish oil, Raynaud’s syndrome, and undiscovered public knowledge. Perspect. Biol. Med. 1986, 30, 7–18.
- Baek, S.H.; Lee, D.; Kim, M.; Lee, J.H.; Song, M. Enriching plausible new hypothesis generation in PubMed. PLoS ONE 2017, 12, e0180539.
- Weeber, M.; Klein, H.; de Jong-van den Berg, L.T.W.; Vos, R. Using concepts in literature-based discovery: Simulating Swanson’s Raynaud–fish oil and migraine–magnesium discoveries. J. Am. Soc. Inf. Sci. Technol. 2001, 52, 548–557.
- Chiang, A.P.; Butte, A.J. Systematic evaluation of drug-disease relationships to identify leads for novel drug uses. Clin. Pharm. 2009, 86, 507–510.
- Andronis, C.; Sharma, A.; Virvilis, V.; Deftereos, S.; Persidis, A. Literature mining, ontologies and information visualization for drug repurposing. Brief. Bioinform. 2011, 12, 357–368.
- Lekka, E.; Deftereos, S.N.; Persidis, A.; Persidis, A.; Andronis, C. Literature analysis for systematic drug repurposing: A case study from Biovista. Drug Discov. Today Ther. Strateg. 2011, 8, 103–108.
- Krassowski, M.; Das, V.; Sahu, S.K.; Misra, B.B. State of the Field in Multi-Omics Research: From Computational Needs to Data Mining and Sharing. Front. Genet. 2020, 11, 610798.
- Batool, M.; Ahmad, B.; Choi, S. A Structure-Based Drug Discovery Paradigm. Int. J. Mol. Sci. 2019, 20, 2783.
- Tan, F.; Yang, R.; Xu, X.; Chen, X.; Wang, Y.; Ma, H.; Liu, X.; Wu, X.; Chen, Y.; Liu, L.; et al. Drug repositioning by applying ‘expression profiles’ generated by integrating chemical structure similarity and gene semantic similarity. Mol. Biosyst. 2014, 10, 1126–1138.
- Kulkarni, J. Selective Estrogen Receptor Modulators-A Potential Treatment for Psychotic Symptoms of Schizophrenia? NCT00361543. Available online: https://clinicaltrials.gov/ct2/show/NCT00361543 (accessed on 28 January 2015).
- Kulkarni, J.; Gavrilidis, E.; Gwini, S.M.; Worsley, R.; Grigg, J.; Warren, A.; Gurvich, C.; Gilbert, H.; Berk, M.; Davis, S.R. Effect of Adjunctive Raloxifene Therapy on Severity of Refractory Schizophrenia in Women: A Randomized Clinical Trial. JAMA Psychiatry 2016, 73, 947–954.
- Henry, L. UMCC 2013.051: Prospective Pilot Study Evaluating the Use of Cyclobenzaprine for Treatment of Sleep Disturbance, Fatigue, and Musculoskeletal Symptoms in Aromatase Inhibitor-Treated Breast Cancer Patients. NCT01921296. Available online: https://clinicaltrials.gov/ct2/show/NCT01921296 (accessed on 21 March 2016).
- Hebbring, S.J. The challenges, advantages and future of phenome-wide association studies. Immunology 2014, 141, 157–165.
- Senthil, G.; Dutka, T.; Bingaman, L.; Lehner, T. Genomic resources for the study of neuropsychiatric disorders. Mol. Psychiatry 2017, 22, 1659–1663.
- Ganapathiraju, M.K.; Thahir, M.; Handen, A.; Sarkar, S.N.; Sweet, R.A.; Nimgaonkar, V.L.; Loscher, C.E.; Bauer, E.M.; Chaparala, S. Schizophrenia interactome with 504 novel protein–protein interactions. NPJ Schizophr. 2016, 2, 16012.
- Karunakaran, K.B.; Chaparala, S.; Ganapathiraju, M.K. Potentially repurposable drugs for schizophrenia identified from its interactome. Sci. Rep. 2019, 9, 12682.
- Gaspar, H.A.; Gerring, Z.; Hübel, C.; Middeldorp, C.M.; Derks, E.M.; Breen, G.; Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium. Using genetic drug-target networks to develop new drug hypotheses for major depressive disorder. Transl. Psychiatry 2019, 9, 117.
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58.
- Shukla, R.; Henkel, N.D.; Alganem, K.; Hamoud, A.-R.; Reigle, J.; Alnafisah, R.S.; Eby, H.M.; Imami, A.S.; Creeden, J.F.; Miruzzi, S.A.; et al. Signature-based approaches for informed drug repurposing: Targeting CNS disorders. Neuropsychopharmacology 2020, 46, 116–130.
- Iorio, F.; Rittman, T.; Ge, H.; Menden, M.; Saez-Rodriguez, J. Transcriptional data: A new gateway to drug repositioning? Drug Discov. Today 2013, 18, 350–357.
- Gaiteri, C.; Ding, Y.; French, B.; Tseng, G.C.; Sibille, E. Beyond modules and hubs: The potential of gene coexpression networks for investigating molecular mechanisms of complex brain disorders. Genes Brain Behav. 2014, 13, 13–24.
- Truong, T.T.; Bortolasci, C.C.; Spolding, B.; Panizzutti, B.; Liu, Z.S.; Kidnapillai, S.; Richardson, M.; Gray, L.; Smith, C.M.; Dean, O.M.; et al. Co-Expression Networks Unveiled Long Non-Coding RNAs as Molecular Targets of Drugs Used to Treat Bipolar Disorder. Front. Pharmacol. 2022, 13, 873271.
- Akbarian, S.; Liu, C.; Knowles, J.A.; Vaccarino, F.M.; Farnham, P.J.; Crawford, G.E.; Jaffe, A.E.; Pinto, D.; Dracheva, S.; Geschwind, D.H.; et al. The PsychENCODE project. Nat. Neurosci. 2015, 18, 1707–1712.
- Hoffman, G.E.; Bendl, J.; Voloudakis, G.; Montgomery, K.S.; Sloofman, L.; Wang, Y.-C.; Shah, H.R.; Hauberg, M.E.; Johnson, J.S.; Girdhar, K.; et al. CommonMind Consortium provides transcriptomic and epigenomic data for Schizophrenia and Bipolar Disorder. Sci. Data 2019, 6, 180.
- Freshour, S.L.; Kiwala, S.; Cotto, K.C.; Coffman, A.C.; McMichael, J.F.; Song, J.J.; Griffith, M.; Griffith, O.L.; Wagner, A.H. Integration of the Drug–Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 2021, 49, D1144–D1151.
- Cabrera-Mendoza, B.; Martínez-Magaña, J.J.; Monroy-Jaramillo, N.; Genis-Mendoza, A.D.; Fresno, C.; Fries, G.R.; Walss-Bass, C.; López Armenta, M.; García-Dolores, F.; Díaz-Otañez, C.E.; et al. Candidate pharmacological treatments for substance use disorder and suicide identified by gene co-expression network-based drug repositioning. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2021, 186, 193–206.
- Rodriguez-López, J.; Arrojo, M.; Paz, E.; Páramo, M.; Costas, J. Identification of relevant hub genes for early intervention at gene coexpression modules with altered predicted expression in schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 98, 109815.
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.-P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease. Science 2006, 313, 1929–1935.
- Vidović, D.; Koleti, A.; Schürer, S.C. Large-scale integration of small molecule-induced genome-wide transcriptional responses, Kinome-wide binding affinities and cell-growth inhibition profiles reveal global trends characterizing systems-level drug action. Front. Genet. 2014, 5, 342.
- Kidnapillai, S.; Bortolasci, C.C.; Udawela, M.; Panizzutti, B.; Spolding, B.; Connor, T.; Sanigorski, A.; Dean, O.M.; Crowley, T.; Jamain, S.; et al. The use of a gene expression signature and connectivity map to repurpose drugs for bipolar disorder. World J. Biol. Psychiatry 2020, 21, 775–783.
- Ben Guebila, M.; Lopes-Ramos, C.M.; Weighill, D.; Sonawane, A.R.; Burkholz, R.; Shamsaei, B.; Platig, J.; Glass, K.; Kuijjer, M.L.; Quackenbush, J. GRAND: A database of gene regulatory network models across human conditions. Nucleic Acids Res. 2022, 50, D610–D621.
- Liu, W.; Tu, W.; Li, L.; Liu, Y.; Wang, S.; Li, L.; Tao, H.; He, H. Revisiting Connectivity Map from a gene co-expression network analysis. Exp. Med. 2018, 16, 493–500.
- Keenan, A.B.; Jenkins, S.L.; Jagodnik, K.M.; Koplev, S.; He, E.; Torre, D.; Wang, Z.; Dohlman, A.B.; Silverstein, M.C.; Lachmann, A.; et al. The Library of Integrated Network-Based Cellular Signatures NIH Program: System-Level Cataloging of Human Cells Response to Perturbations. Cell Syst. 2018, 6, 13–24.
- Dolmetsch, R.; Geschwind, D.H. The human brain in a dish: The promise of iPSC-derived neurons. Cell 2011, 145, 831–834.
- Huang, J.K.; Carlin, D.E.; Yu, M.K.; Zhang, W.; Kreisberg, J.F.; Tamayo, P.; Ideker, T. Systematic Evaluation of Molecular Networks for Discovery of Disease Genes. Cell Syst. 2018, 6, 484–495.e485.
- Zhang, Z.; Zhang, J. A Big World Inside Small-World Networks. PLoS ONE 2009, 4, e5686.
- Kauppi, K.; Rosenthal, S.B.; Lo, M.-T.; Sanyal, N.; Jiang, M.; Abagyan, R.; McEvoy, L.K.; Andreassen, O.A.; Chen, C.-H. Revisiting Antipsychotic Drug Actions Through Gene Networks Associated With Schizophrenia. Am. J. Psychiatry 2018, 175, 674–682.
- De Bastiani, M.A.; Pfaffenseller, B.; Klamt, F. Master Regulators Connectivity Map: A Transcription Factors-Centered Approach to Drug Repositioning. Front. Pharmacol. 2018, 9, 697.
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082.
- Whirl-Carrillo, M.; Huddart, R.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Whaley, R.; Klein, T.E. An Evidence-Based Framework for Evaluating Pharmacogenomics Knowledge for Personalized Medicine. Clin Pharm. 2021, 110, 563–572.
- Kuhn, M.; Campillos, M.; Letunic, I.; Jensen, L.J.; Bork, P. A side effect resource to capture phenotypic effects of drugs. Mol. Syst. Biol. 2010, 6, 343.
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368.
- Zhou, M.; Wang, Q.; Zheng, C.; John Rush, A.; Volkow, N.D.; Xu, R. Drug repurposing for opioid use disorders: Integration of computational prediction, clinical corroboration, and mechanism of action analyses. Mol. Psychiatry 2021, 26, 5286–5296.
- Goh, K.-I.; Cusick Michael, E.; Valle, D.; Childs, B.; Vidal, M.; Barabási, A.-L. The human disease network. Proc. Natl. Acad. Sci. USA 2007, 104, 8685–8690.
- Lüscher Dias, T.; Schuch, V.; Beltrão-Braga, P.C.B.; Martins-de-Souza, D.; Brentani, H.P.; Franco, G.R.; Nakaya, H.I. Drug repositioning for psychiatric and neurological disorders through a network medicine approach. Transl. Psychiatry 2020, 10, 141.
- Langhauser, F.; Casas, A.I.; Dao, V.-T.-V.; Guney, E.; Menche, J.; Geuss, E.; Kleikers, P.W.M.; López, M.G.; Barabási, A.-L.; Kleinschnitz, C.; et al. A diseasome cluster-based drug repurposing of soluble guanylate cyclase activators from smooth muscle relaxation to direct neuroprotection. NPJ Syst. Biol. Appl. 2018, 4, 8.
- Csermely, P.; Korcsmáros, T.; Kiss, H.J.M.; London, G.; Nussinov, R. Structure and dynamics of molecular networks: A novel paradigm of drug discovery: A comprehensive review. Pharmacol. Ther. 2013, 138, 333–408.
- Chen, Y.-W.; Diamante, G.; Ding, J.; Nghiem, T.X.; Yang, J.; Ha, S.-M.; Cohn, P.; Arneson, D.; Blencowe, M.; Garcia, J.; et al. PharmOmics: A species- and tissue-specific drug signature database and gene-network-based drug repositioning tool. iScience 2022, 25, 104052.
- Sadegh, S.; Skelton, J.; Anastasi, E.; Bernett, J.; Blumenthal, D.B.; Galindez, G.; Salgado-Albarrán, M.; Lazareva, O.; Flanagan, K.; Cockell, S.; et al. Network medicine for disease module identification and drug repurposing with the NeDRex platform. Nat. Commun. 2021, 12, 6848.
- Chen, Y.; Elenee Argentinis, J.D.; Weber, G. IBM Watson: How Cognitive Computing Can Be Applied to Big Data Challenges in Life Sciences Research. Clin. Ther. 2016, 38, 688–701.
More