The main challenge in diagnosing and managing thoracic aortic aneurysm and dissection (TAA/D) is represented by the early detection of a disease that is both deadly and “elusive”, as it generally grows asymptomatically prior to rupture, leading to death in the majority of cases. Gender differences exist in aortic dissection in terms of incidence and treatment options. Efforts have been made to identify biomarkers that may help in early diagnosis and in detecting those patients at a higher risk of developing life-threatening complications. As soon as the hereditability of the TAA/D was demonstrated, several genetic factors were found to be associated with both the syndromic and non-syndromic forms of the disease, and they currently play a role in patient diagnosis/prognosis and management-guidance purposes.
- thoracic aortic aneurysm and dissection
- syndromic aortopathies
- differential diagnosis
- genetics
- biomarkers
- genetic diagnosis
1. Introduction
2. Drivers of TAA Formation: A Constant Journey through Gene Discovery
The complexity and heterogeneity of TAA characteristics, syndromic presentation, and/or progression is the consequence of multiple but unique cellular and molecular-genetic mechanisms underlying its development, which often result in similar clinical presentation [14]. As familial-aggregation studies have suggested, more than 20% of patients have at least one first-degree family member with an arterial aneurysm, basically defining an increased risk for relatives of the affected individuals [15]. The first clue about the heritability of the trait is derived from case-control studies comparing the prevalence of thoracic aortic aneurysms, thoracic aortic dissections, and sudden death in first-degree relatives of patients referred for thoracic aortic surgery [16], identifying a higher risk for developing those diseases in the proband first-degree relatives with respect to the control groups (with relative risks of 1.8, 10.9, and 1.8 in proband fathers, brothers, and sisters, respectively). More evidence on genetic factors contributing to TAA development was provided by an analysis of a database comprising 598 patients evaluated for TAA in the United States [17], which showed a faster growth rate of aortic aneurysm in patients with familial cases with respect to the sporadic ones, with a younger age of presentation. In addition, pedigrees also showed different patterns of inheritance (autosomal dominant, X-linked, autosomal recessive). The role of genetic factors in causing TAA was further confirmed by more recent studies as well, analyzing different type of aneurysms [18]. It was mainly through genetic and animal models’ studies that the combination of disrupted/altered cellular processes driving the TAA formation were elucidated as well as the specific associated genes (Table 1). In this regard, it has to be noticed, which some causative genes exert an overlapping effect on both syndromic and non-syndromic TAAs, even if these two conditions have traditionally been considered as distinct entities (e.g., ACTA2, SMAD3) (Table 1) [6].| Biological Process/Cellular Compartment | Gene | Protein | OMIM | Syndromic TAA/D | Non-Syndromic FTAA/D | Associated Syndrome/Diseases |
|---|---|---|---|---|---|---|
| Extracellular matrix/remodeling | BGN | Biglycan | 300,989 | + | − | Meester-Loeys syndrome. ARD, TAAD, pulmonary artery aneurysm, IA, arterial tortuosity [19]. |
| COL3A1 | Collagen Type III α1 Chain | 130,050 | + | − | EDS, vascular type IV. TAAD, early aortic dissection, visceral arterial dissection, vessel fragility [20]. | |
| EFEMP2 | EGF Containing Fibulin Extracellular Matrix Protein 2 | 614,437 | + | − | Cutis laxa, AR type Ib. Ascending aortic aneurysms, other arterial aneurysms, arterial tortuosity, stenosis [21]. | |
| ELN | Elastin | 123,700 185,500 |
+ | − | Cutis laxa. AD ARD, ascending aortic aneurysm and dissection [22], TAA [23][24], BAV, IA possibly associated with SVAS. | |
| FBN1 | Fibrillin-1 | 154,700 | + | + | Marfan syndrome. ARD, TAA [25], TAAD [26], AAA, other arterial aneurysms, pulmonary artery dilatation, arterial tortuosity [27]. | |
| LOX | Protein-lysine 6-oxidase | 617,168 | − | + | AAT10. AAA, hepatic artery aneurysm, BAV, CAD, TAAD [28][29]. | |
| MFAP5 | Microfibril Associated Protein 5 | 616,166 | − | + | AAT9. ARD, TAA [30][31]. | |
| Smooth muscle cells | ACTA2 | Smooth muscle α-actin | 611,788 613,834 614,042 |
+ | + | AAT6, multisystemic smooth muscle dysfunction, MYMY5. Early aortic dissection, CAD, stroke (moyamoya disease), PDA, pulmonary artery dilation, BAV, TAAD, TAA [24][32]. |
| FLNA | Filamin A | 300,049 | + | − | Periventricular nodular heterotopia and otopalatodigital syndrome. Aortic dilatation/aneurysms, peripheral arterial dilatation, PDA, IA, BAV, TAA [32][33]. | |
| MYH11 | Smooth muscle myosin heavy chain | 132,900 | − | + | AAT4. PDA, CAD, peripheral vascular occlusive disease, carotid IA, TAAD, early aortic dissection [32][34][35]. | |
| MYLK | Myosin light chain kinase | 613,780 | − | + | AAT7. TAAD, early aortic dissections [36][37]. | |
| TGF-β signaling | LTBP1 | Latent TGF-β binding protein 1 | 150,390 | + | − | Aortic dilation with associated musculoskeletal findings. Dental anomalies, short stature. TAAD, AAA, visceral and peripheral arterial aneurysm [38]. |
| LTBP3 | Latent TGF-β binding protein 3 | 602,090 | ||||
| SMAD2 | SMAD2 | 619,657 619,656 |
+ | - | Unidentified CTD with arterial aneurysm/dissections. ARD, ascending aortic aneurysms, vertebral/carotid aneurysms and dissections [39], AAA. | |
| SMAD3 | SMAD3 | 613,795 | + | + | LDS type III. ARD, TAAD [40], early aortic dissection [39], AAA, arterial tortuosity, other arterial aneurysms/dissections [9], IA, BAV. | |
| SMAD4 | SMAD4 | 175,050 | + | - | JP/HHT syndrome. ARD, TAAD [39], AVMs, IA. | |
| SMAD6 | SMAD6 | 602,931 | - | + | AOVD2. BAV/TAA [24]. | |
| TGFB2 | TGF-β2 | 614,816 | + | + | LDS type IV. ARD, TAA [40], TAAD, arterial tortuosity [39], other arterial aneurysms, BAV. | |
| TGFB3 | TGF-β3 | 615,582 | + | - | LDS type V. ARD, TAAD, AAA/dissection, other arterial aneurysms, IA/dissection [39]. | |
| TGFBR1 | TGF-β receptor type 1 |
609,192 | + | + | LDS type I+AAT5. TAAD [40], early aortic dissection, AAA, arterial tortuosity, other arterial aneurysms/dissection [9], IA, PDA, BAV. | |
| TGFBR2 | TGF-β receptor type 2 |
610,168 | + | + | LDS type II+AAT3. TAAD [40], early aortic dissection, AAA, arterial tortuosity, other arterial aneurysms/dissection [9], IA, PDA, BAV. | |
| Others | AXIN1/PDIA2 locus | − | − | + | − | BAV. BAV/TAA [41]. |
| FBN2 | Fibrillin-2 | 121,050 | + | − | Contractual arachnodactyly. Rare ARD and aortic dissection [42], BAV, PDA. | |
| FOXE3 | Forkhead box 3 | 617,349 | − | + | AAT11. TAAD [30] (primarily type A dissection). | |
| MAT2A | Methionine adenosyl-transferase II α | n.a. | − | + | FTAA Thoracic aortic aneurysms [30][43]. BAV. | |
| NOTCH1 | NOTCH1 | 109,730 | − | + | AOVD1. BAV/TAAD [24]. | |
| PRKG1 | Type 1 cGMP-dependent protein kinase | 615,436 | − | + | AAT8. TAAD [28][43], early aortic dissection, AAA, coronary artery aneurysm/dissection, aortic tortuosity, small vessel, CVD. | |
| ROBO4 | Roundabout guidance receptor 4 | 607,528 | − | + | BAV. BAV/TAA [24]. | |
| SKI | Sloan Kettering proto-oncoprotein | 182,212 | + | − | Shprintzen–Goldberg syndrome. ARD, arterial tortuosity, pulmonary artery dilation, other (splenic) arterial aneurysms [36]. | |
| SLC2A10 | Glucose transporter 10 | 208,050 | + | − | Arterial tortuosity syndrome. ARD, ascending aortic aneurysms [36], other arterial aneurysms, arterial tortuosity [44], elongated arteries, aortic/pulmonary artery stenosis. |
2.1. Extracellular Matrix Components
Among the genes causing and/or influencing TAA development, those codifying the ECM components are always mentioned first due to the amount of data that was collected over the years through animal studies, providing evidence of their impact on maintaining the structural integrity of the aortic wall. Those components are in close relationship and represent key factors, upstream/downstream, and intermediate elements of cellular pathways with impairment that has been demonstrated to have consequences in aneurysm development/predisposition in different ways, namely depletion of the elastic lamina in the aortic wall, lengthening of the ascending aorta, impaired assembly of collagen and elastic fibers, and altered TGF-β signaling (Figure 1).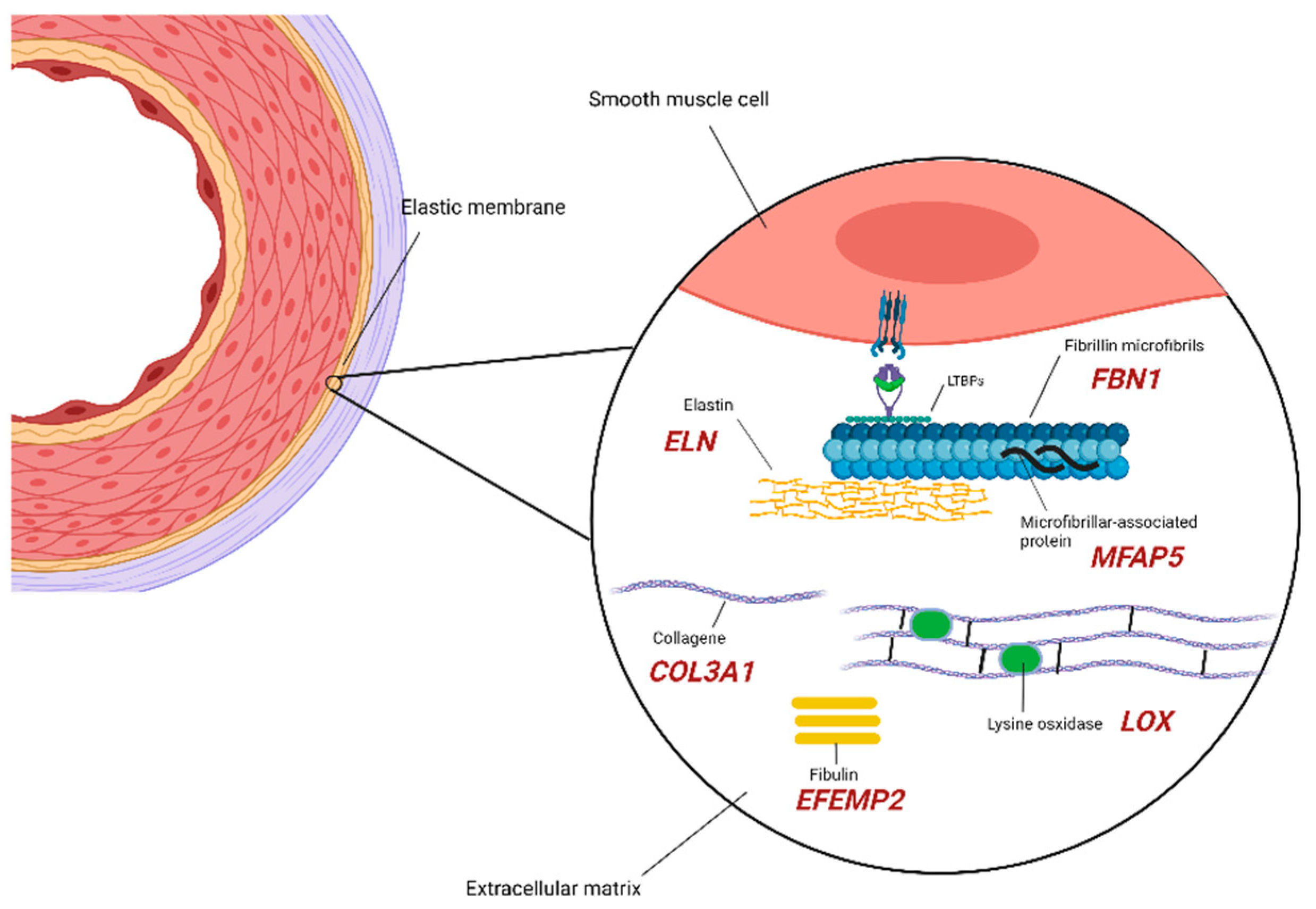
2.2. SMCs (Smooth Muscle Cells) Compartment
The aorta is, for the most part, constituted by populations of SMCs, and the maintenance of the contractile properties of their cellular components is highly controlled and regulated. Molecular studies have, in fact, demonstrated that the impairment at different levels of this strictly regulated system predispose individuals to TAA development. Among proteins participating in this cellular machinery, α-smooth muscle aorta encoded by ACTA2 gene (10q23.31) exerts an important function in maintaining contractility, and its depletion is, in turn, associated with a decrease ability of the monomer to assemble into polymeric filaments, eventually impairing the actin-myosin contractile unit [54]. Decades of study has proven mutations in this gene represent an important cause of familial and nonfamilial non-syndromic TAAD [55][56][57], accounting for 12–21% of TAAD. Filamin A (FLNA gene, Xq28) is an anchoring cytoskeletal protein that, among its diverse functions, acts as a linker between actin filaments [58]. An 18.4% TAA frequency was found in a large systematic analysis of both pediatric and adult patients with periventricular nodular heterotopia carrying loss-of-function FLNA mutations [33] and, very recently, in a patient developing TAA with an history of systemic lupus erythematosus [59]. Myosin heavy chain 11 (MYH11 gene, 16p13.11) interacts with α-actin, controlling its state change and enabling the actin filaments binding. MYH11 is also traditionally classified as a mostly non-syndromic gene for TAA and was demonstrated to be associated with other manifestations such as aortic stiffness, an early hallmark of the disease [34][60]. However, it has to be noticed that MYH11 defects account for <1% of all non-syndromic TAAD, with an aorta diameter prior to dissection usually >5 cm [35]. Robust data support the idea of shared defects in FLNA, MYH11, and ACTA2 machinery, determining mechanical-strength disruption of the vascular walls and contractility-maintenance failure as significant causes of TAA progression, especially in non-syndromic cases [32]. The MYLK gene (3q21.1) encodes the myosin light chain kinase, regulating the actin-myosin interaction and phosphorylating myosin light-chains, and its most destructive mutations were once again mostly found in non-syndromic TAAD [9] (Figure 2).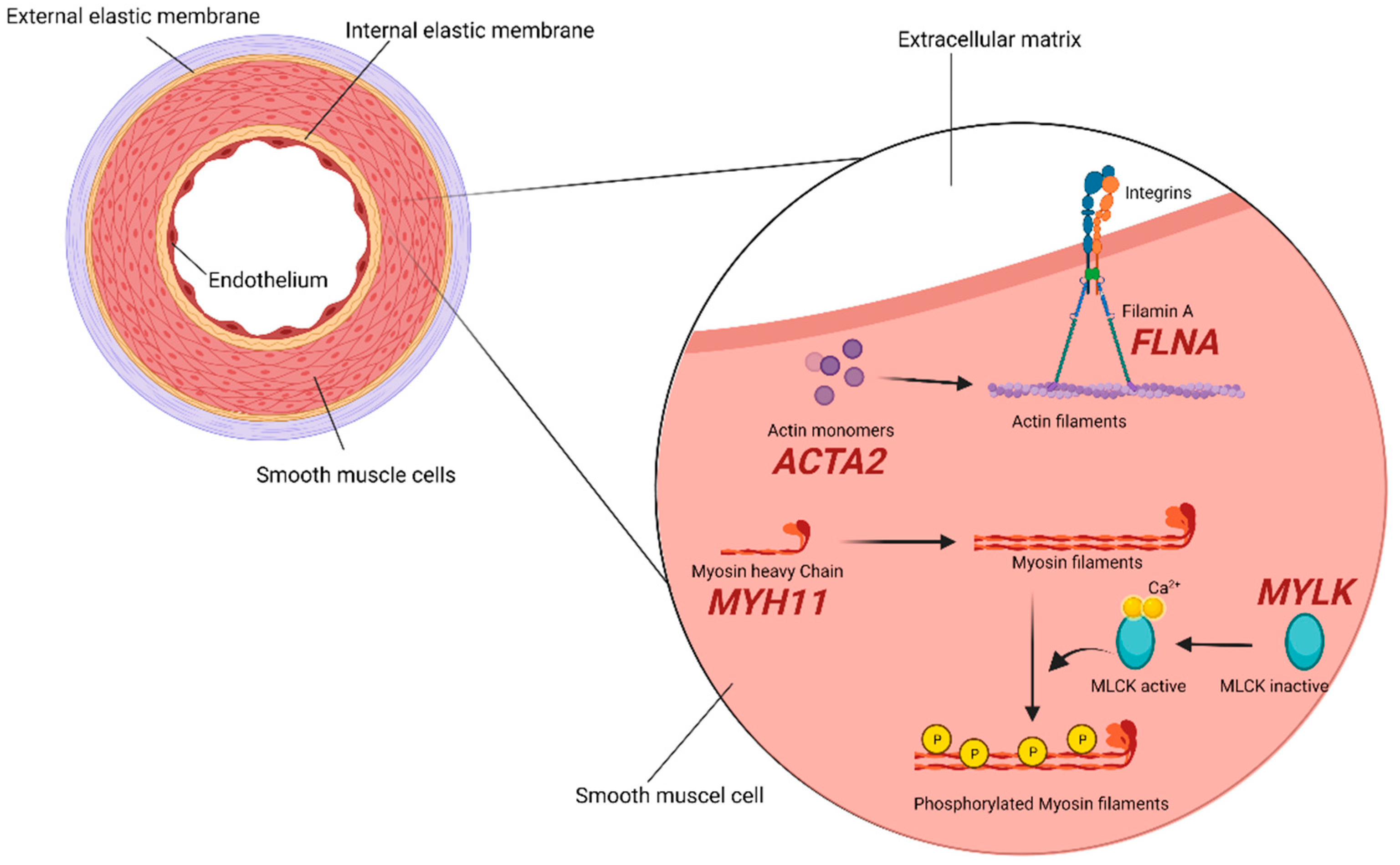
2.3. TGF-β Signaling
A mutational repertoire in the genes coding for positive and negative regulators of TGF-β signaling has been reported in patients developing TAA in association with MFS and LDS, with those large volumes of animal studies research representing the first, and one of the major, contributions to researchers' understanding of aneurysmal onset and growth [61]. TGF-β signaling, in fact, plays a critical role in a series of vascular cellular processes such as blood-vessel development and maintenance, positive regulation of contractile proteins’ expression, cell differentiation, proliferation, and homeostasis, and those mechanisms have been demonstrated to be dysregulated in all types of LDSs [61]. Different models have been proposed as the basis of aortic-wall dilatation, one of those suggesting a reduction in TGF-β signaling that would cause an impaired expression of contractile proteins, resembling the effect of mutations in ACTA2 and MYH11 genes [62]. Alternatively, LDS-causing mutations increase the VSMCs’ signaling capacity, resulting in a defective responsiveness to the TGF-β of cardiac neural crest-derived VSMCs, which are highly abundant in the proximal thoracic aorta and in excessive activation of TGF-β signaling [63][64]. TGF-β is secreted by many types of cells, including macrophages, as part of a large latent complex that consists of the mature TGF-β cytokine, a dimer of its processed latency associated peptide (LAP), and one of three latent TGF-β binding protein-isoforms (LTBP1, 3, or 4). The latter binds to ECM components such as fibronectin or microfibrils composed of fibrillin-1. Among the LTBP protein family, a deletion involving the LTBP1 gene (2p22.3) has been found to segregate in a three-generation family presenting with TAA [38], while a variant involving LTBP3 gene (11q13.1) was suggested to be predisposing to TAA/D development [65]. Upon release, TGF-β binds to its heterodimeric receptor, activating the phosphorylation of the SMAD2 and SMAD3 proteins, which transmit the signal to the nucleus via the association with SMAD4 and, in turn, activate gene transcription [66]. Upon LDS-causing mutations, those affecting elements of the TGF-β signaling pathways involve the receptor heterodimer, made of the two components codified by the TGFBR1 (9q22.33) and TGFBR2 (3p24.1) genes; …these mutations result in decreased kinase activity with a reduction in the transduction molecules levels codified by the SMAD2 (18q21.1) and SMAD3 (15q22.33) genes [46]. The SMAD2 and SMAD3 proteins belong, in fact, to the receptor-activated (R)-SMAD family, intracellular effectors of the canonical TGF-β signaling pathway, with activated ligands that include the TGF-β2 and TGF-β3 encoded by TGFB2 (1q41) and TGFB3 (14q24.3). Mutations involving those genes have been associated with different subtypes of LDS, sharing aneurysm formation as a common clinical manifestation and being characterized by the presence/absence of other systemic features such as aortic or arterial tortuosity, cleft palate, bifid uvula, mitral valve disease, skeletal overgrowth, and so on [39] as well as a different tendency to early aortic dissection. TGFBR1, TGFBR2 and SMAD3 mutations also account for up to 3%, 5% and 2%, respectively, of non-syndromic FTAAD [32] (Figure 3).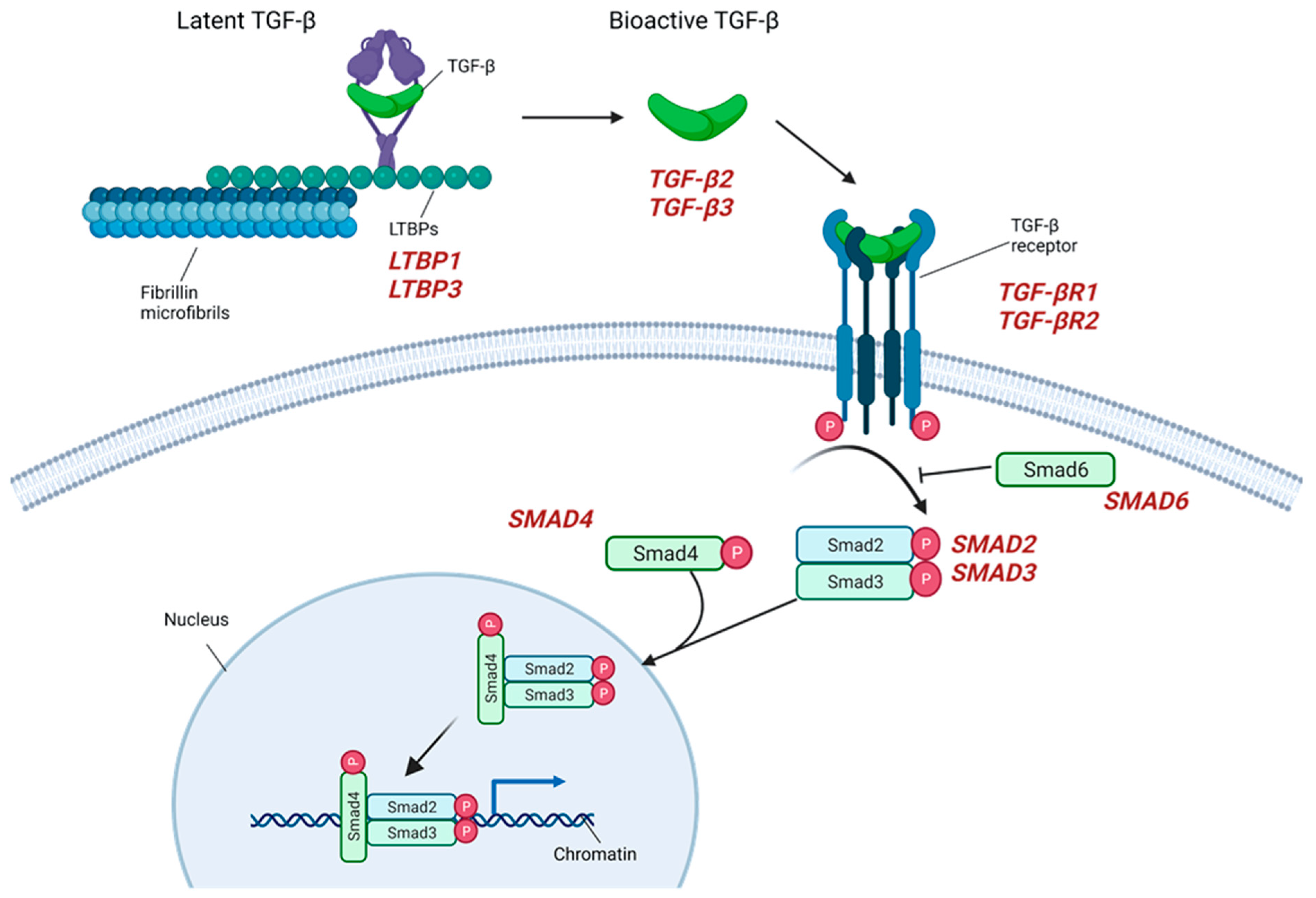
2.4. TAA in the Context of Bicuspid Aortic Valve and the Role of Proteases
Since approximately 40% of BAV patients are prone to develop ascending aortic dilatation, this congenital heart defect is currently considered as an independent risk factor for TAA [7][67]. As the two phenotypes frequently occur together, and given the autosomal pattern of inheritance with incomplete penetrance that has been proposed for BAV, hypotheses have been made about the pathophysiological mechanisms driving the valve anomaly, along with its more frequent complication that, among others, involves the potential role of genetic syndromic TAA-associated variants such as NOTCH1, ROBO4, SMAD6, ELN, FBN1, ACTA2, and LOX [24][68]. Metalloproteases (MMPs) have been hypothesized to participate in TAA development in the context of BAV, following the observation of a significant increase in MMP-2 levels associated with a reduction in TIMP-1 in BAV/TAA compared with TAA subjects, in the context of a normal tricuspid valve; a greater activity of MMP-2 and MMP-9 in aneurysms was associated with BAV, which could explain the higher prevalence of TAA in these patients [69][70]. Apart from BAV, the combination of altered levels of MMPs, ADAMTS, and TIMPs (the main proteases and inhibitors within the media controlling the ECM environment’s integrity and maintenance) are proven to actively contribute to the medial degeneration triggering TAA [71]. Studies on human and animal models led to the observation of increased expression of MMP-2 and MMP-9 in TAA intima and media [72] and higher levels of ADAMTS-1 and ADAMTS-4 in sporadic, ascending TAA tissues from human patients [73]. Despite the indisputable value of these observation in understanding TAA pathogenesis and their potential implications in diagnosis, it has to be noticed that those markers are not specific for the phenotype, and their levels are altered in several other processes such as AAA (MMP-2, MMP-9), cancer (MMP-9, TIMP-1, TIMP-2), renal disease (TIMP-2), and so on [74] and are currently not included in the recommended work-up for TAA diagnosis and management nor in the genetic screening.3. Mechanisms of TAA Progression: The Dissection Menace
As the pathophysiological mechanisms underlying TAA onset in both its syndromic and non-syndromic presentation have been illustrated, and their specific molecular features continue to be unraveled thanks to the advancements in wide genetic-screening technologies, detection of the disease remains a clinical challenge. This is especially relevant with respect to its most catastrophic complications, rupture and dissection, leading to death in the great majority of patients without timely treatment [4][75]. In fact, once aortic dissection has occurred, mortality is 1–2% for each hour afterwards, resulting in a 48-h mortality of approximately 50% [76]. However, in the case of survival, serious complications may follow such as lethal malperfusion syndrome, aortic regurgitation, cardiac failure, and stroke [77]. Dissection represents a considerable diagnostic challenge for physicians due to the rarity of the condition and the characteristic symptomatology often mimicking other, more common diseases, determining a delay in diagnosis in >30% of cases [4]. Thus, the understanding of the pathophysiology, the key features, and the potential biochemical/molecular markers of TAA progression into aortic dissection has been crucial during the last decades to improve outcomes, for long-term prognosis, and eventually for patients’ risk-stratification purposes.3.1. Pathophysiology and Risk Factors
The instability and the deteriorating integrity of the aortic wall may be due to being predisposed to inherited conditions (such as inherited connective tissue disorders) or can be acquired, as happens with atherosclerotic degeneration due to ageing. Two mechanisms have been proposed to initiate the dissection cascade: (1) in most cases, a tear in the intima exposes the medial layer to the pulsatile blood flow; and (2) in fewer cases, the rupture of the vasa vasorum leads to the weakening of the inner aortic wall [78]. In the first scenario, a false lumen derives from the progressive separation of the aortic wall layers, and its propagation leads to aortic rupture where the adventitia is disrupted: rupture quickly leads to exsanguination and death [79]. In the latter case, the bleeding results in intramural hematoma that may progress in aortic dissection. It has actually been hypothesized that a co-existence of these two conditions may, in turn, constitute a spectrum [80]. From a molecular point of view, dissection occurs as a consequence of the aortic-wall structure’s remodeling, due to inflammation and ECM-degradation processes. Once again, proteases exert an important role, since the infiltration of the activated macrophages and pro-inflammatory cytokines in the tunica media leads to an excessive production of MMP-1, MMP-9, and MMP-12 and to an imbalance between them and their inhibitors (TIMPs), which, in turn, results in the degradation of collagen and elastin fibers [81][82]. Wall remodeling is also maintained by VEGF-mediated neo-angiogenesis, as the production of VEGF (also functioning as a pro-inflammatory molecule) is increased in degraded medial layers [83]. Among classical risk factors associated with aortic dissection, namely older age, dyslipidemia, and increased levels of apolipoprotein A1, 80% of patients developing dissection have hypertension [84], which has a direct effect on the pathogenic mechanisms described above. Specifically, hypertension is demonstrated to promote a pro-inflammatory environment mainly by inducing macrophage recruitment and activation [85]; hypertensive patients, in fact, show high concentrations of VEGF, IL-6, MMP-2, and MMP-9 [86][87]. Other risk factors are recognized such as the male sex, a smoking habit, and the concurrence of connective tissue disorders such as MFS, LDS, vEDS, and BAV. Aortic dilatation is known to increase the risk of dissection, with the incidence complications reaching 30% at diameters > 60 mm [88]. Still, dilatation is proven not to be essential for developing a dissection, as ~60% of non-syndromic type A aortic dissection have diameters < 55 mm, while, in the absence of hypertension, MFS or BAV patients show a tendency to dissect at larger diameters [89][90]. Rare risk factors for aortic dissection are further represented by vascular inflammation due to autoimmune disorders such as Giant-cell arteritis, Takayasu Arteritis, Behçet disease, and systemic lupus erythematosus, while 1% to 5% of aortic dissections are secondary to aortitis [91].3.2. Genetic Profiles of Dissection
To date, more than 30 TAAD-causative genes have been discovered (Table 1) and in the context of risk-prediction of TAA progression into potentially fatal dissection, it appears of primary interest to identify those genes or the specific type of variants/genetic profiles that, among others, are more prone to trigger and drive those processes eventually leading to sudden aortic-wall rupture. Marked in bold in Table 1, those genes were demonstrated to increase the risk of dissection at certain aortic sizes. Some of them represent causative genes of peculiar connective-tissue disorders described in association with TAA in its syndromic presentation. Pathogenic variants in FBN1, the MFS-causative gene, were found to increase the risk for Stanford type A and B dissection, even in the context of a normal or minimally dilated ascending aorta [92][93][94]. Indeed, a diagnosis of MFS is established in ~5% of patients with aortic dissection [95]. Haploinsufficiency, mainly resulting from truncating or splicing FBN1 mutations, is described as the leading mechanism behind the increased rate of aortic events [27]. Some overlapping cardiovascular clinical manifestations characterize MFS and LDS as basically reflecting a predisposition of patients affected by one or the other connective disorder to develop aneurysm and dissections of aorta and other arteries [92]. Implicated genes are TGFBR1, TGFBR2, and SMAD3, causing LDS type I and III, which have been associated with increased aortic risk of dissection at diameters < 50 mm [9]. TGFBR1 and TGFBR2 mutations’ carriers are often reported as a comparable clinical picture regarding presentation and natural history, even though clinical differences have actually been observed between the two populations of mutated subjects [96]. Regarding TGFBR1 families, a gender-based difference in survival has been observed, resulting in significantly better outcomes in women than in men. Differences were also observed in terms of aortic diameter, with TGBFR2 carriers dissecting at minimal aortic dilatation with respect to TGFBR1 carriers in which the ascending aorta diameter at the time of type A dissections was 50 mm. A more recent and large multicenter retrospective registry of patients with genetically triggered thoracic aortic disease reported the data of 441 subjects harboring mutations on the TGF-β receptor genes, somewhat confirming the previous observations in TGFBR2 mutations’ female carriers, that type A dissections of moderately dilated ascending aorta appeared more frequently than in males, which was not the case with TGFBR1, suggesting a more aggressive aortic disease in TGFBR2 patients, especially in women [97]. Together with a TGFBR2 mutation and the female sex, other features such as aortic tortuosity, hypertelorism, and translucent skin were found to be associated with an increased aortic dissection risk and may be taken into consideration in determining the optimal surgical timing (45 mm in the general population, lowered toward 40 in females with low body surface area, harboring a TGFBR2 mutation, and presenting extra-aortic features). The literature data support the role of SMAD3 as a dissection-predisposing gene [66][98][99][100], with mutations’ carriers having a cumulative risk of dissection or prophylactic surgical repair of 50% by age 50 and 85% by age 80. Interestingly, these subjects are characterized by a later onset of aortic events, possibly leading to a delayed diagnosis if compared, for instance, to MFS patients presenting with wider systemic features [101]. The early recognition of the disease, in this subset of patients, consequently lies in the family history of thoracic aortic disease (as a key element), so this observation remarks on the need to identify those subjects before dissection occurs. The majority (63%) of SMAD3 mutations are missense and reside in the MH2 domain, which regulates the oligomerization with SMAD2 or SMAD4 and the subsequent activation of transcription, with those variants being associated with earlier aortic events compared to truncating, non-sense, gene-disrupting ones [102]. Even if generally associated with a specific connective-tissue disorder, it must be reasserted that FBN1, TGFBR1, TGFBR2, SMAD3, and TGFB2 mutations also account for an additional 14% of non-syndromic familial TAA [40], in which the diagnosis of aortic disease may be complicated by the absence of peculiar systemic clinical features. A vEDS-causing gene, COL3A1, is also among those referenced as dissection associated ones, due to the supporting literature data on population studies and case reports. The dissection was found to develop in different locations of the arterial tree, such as the abdominal aorta, as well as the iliac, coronary, and cervical arteries [103][104][105][106]. Concerning syndromic TAA in patients diagnosed as vEDS, few post-mortem cases were reported in 2010 [107], and 33 unrelated individuals or families were found to carry COL3A1 splicing mutations or small deletions partially removing splice-junctions sequences [108], and patients developing post-surgical or sudden aortic events are reported [109][110][111]. COL3A1 variants were additionally found to be associated with sporadic forms of TAAD in recent WES and case-control studies [112][113]. The type of variant involving the COL3A1 gene was also been suggested to correlate with the phenotype severity of vEDS; specifically, a subgroup of patients in a large European cohort bearing non-glycine missense and/or genetic variations at the C- and N-termini of type III procollagens was found to develop a later-onset and a milder phenotype with higher rates of aortic complications [114], while mutations at splice-donor sites were associated with higher mortality rates with respect to those involving the splice-acceptor sequences [115]. The literature data on animal models/human cohorts have identified a number of other genes, with mutations that were suggested to increase the risk of aortic dissection, such as EFEMP2 at the level of the ascending aorta [21], MYH11, ACTA2, and MAT2A in the thoracic aorta [43][116], and SLC2A10 at the aorta, as well as the arteries [44] LOX and PRKG1, with more limited data [28][117], and FOXE3 and MFAP5 as identified through WES studies [30][31].4. Genetic Testing in Supporting TAA/D Diagnostics and In Risk Prediction: Where Do We Stand?
Although primarily considered as surgical disease, TAA’s optimal management greatly relies on an appropriate workup with the major purpose of identifying those features suggestive of a rapid progression of the aortic anomaly, thus predicting potentially life-threatening consequences of the disease. In this context, an accurate genetic evaluation/diagnosis serves different purposes: (a) guidance for overall medical management and surgical options; (b) timely evaluation of other organs that could be affected essentially in syndromic forms of TAA; (c) better definition of the prognosis; (d) identification of high-risk first-degree family members; (e) estimation of recurrence risk for future pregnancies in the prenatal diagnosis’ framework; and (f) support for imaging techniques in capturing nonsyndromic TAA patients who may be missed while developing dissection or rupture before reaching the guidelines-defined aortic diameter thresholds for aortic intervention [[118]]. As previously mentioned, syndromic and non-syndromic heritable thoracic aortic disease are, in most cases, inherited in an autosomal dominant manner except for rare X-linked and recessive conditions [[119]]. The accurate clinical evaluation of at-risk relatives is critical in this context, and ordinary and reproductive pre- and post-test genetic counseling allow for the early identification of an undiagnosed aortic disease in the first case and provide awareness about the risk of transmission to the offspring in the latter. Mutations are described to have variable penetrance depending on the TAA presentation, from almost 100% in MFS and 90% in LDS, to 50% in FTAAD and BAV in the presence of ascending aortic aneurysm. In fact, in the case of FTAAD, the causal mutation is found in much fewer cases (<10%) than in MFS or LDS, this discrepancy also being evident at the phenotypic level, presenting with a different severity of clinical manifestations along with age of presentation or diagnosis. When features of a connective-tissue disorder are present, patients should undergo genetic counselling and testing where appropriate [[10]]. The current ESC guidelines recommend genetic screening in first-degree relatives of TAA or aortic dissection and a diagnosis of familial aortic disease. In absence of a genetic diagnosis, at-risk relatives should undergo examination every 5 years. Screening should cover the entire arterial tree (including cerebral arteries) in families with nonsyndromic familial aortic disease [[120]]. According to the North American guidelines and related Class I recommendations, in case of identification of a mutation in one of the following genes, FBN1, TGFBR1, TGFBR2, COL3A1, ACTA2, and MYH11, which are associated with aortic aneurysm and/or dissection, first-degree relatives should undergo counseling and testing. Then, only the relatives with the genetic mutation should undergo aortic imaging. The guidelines provide some more recommendations (Class IIa and IIb): (a) ACTA2 sequencing should be considered in case of family history of thoracic aortic aneurysm and/or dissection; (b) TGFBR1, TGFBR2, and MYH11 sequencing may be considered in patients with a family history and clinical features associated with mutations in these genes; and c) if one or more first-degree relatives of a patient with known thoracic aortic aneurysm and/or dissection are found to have thoracic aortic dilatation, aneurysm, or dissection, then referral to a geneticist may be considered [[121]]. Following the exclusion of a syndromic condition, nonsyndromic TAA, in which mutations in genes known to be involved in syndromic forms of TAAD are rarely found, may present suggestive features of a genetic etiology, which might include young age at presentation (<50 years old), multiple aneurysms or dissections, and aortic root aneurysm [[122],[123]]. In this scenario, genetic counseling should begin with the collection of the most detailed information of a three-generation family history, for the presence of aneurysm, dissection, sudden deaths, and syndromic features that would help in determining the inheritance pattern, identifying at-risk relatives, and recognizing syndromic signs [[119]]. In 2009, Ripperger and co-workers reported three cases of sudden, unexpected death due to thoracic aortic dissection, pointing out the great benefit that could be derived from alerting the at-risk relatives of the deceased about a potential heritable etiology of the disease [[124]]. The authors propose the development of a standard procedure which includes genetic counseling for at-risk relatives and storage of DNA or unfixed tissue for molecular investigations that would eventually allow differential diagnostic reappraisal from a genetic point of view. In any case, during genetic consultation, patients should become aware of the limitations, benefits, and personal and familial implications of genetic testing. Besides, awareness should be raised on the possibility of a negative genetic test that would not necessarily exclude a genetic etiology, thus indicating the imaging to be performed anyways in the first-degree family members in the search for aortic disease [[121]]. In fact, some types of genetic variants may be undetectable by standard assays and, similarly, the causative mutation may involve a gene that has not yet been associated to TAAD, due to absence of data supporting the actual pathological effect of that variant [[121]]. As a matter of fact, regarding the most appropriate genetic test selection, no specific indications are provided by the European guidelines. Genetic-testing panels vary significantly among laboratories and despite the enthusiasm for the so-called “exome-first” approach in diagnosing such a complex disease as TAA, its actual benefit and routine application in the diagnostic workup currently represent a matter of debate within the international scientific community.
5. Take home message
TAA’s bad reputation of “silent killer” is to be ascribed to its characteristic features, including its slow and gradual formation and the absence of visible signs, with patients remaining asymptomatic. This condition is elusive and yet potentially life-threatening, as it manifests itself only once the aneurysm is large enough to lead to an acute and devastating aortic event, with a significant percentage of patients dying before reaching the hospital. As a result, it is of the utmost relevance to identify biomarkers for the early identification of asymptomatic patients, a task which is both essential and challenging. In this regard, there’s an important distinction to be made between the TAD management within the emergency department, in which the room to maneuver is objectively limited, and those other situations in which the fatal event has not happened yet. In the first case, as Mehta and co-workers pointed out in a very recent review, the margin of intervention is essentially directed to improving the patient’s outcome by different means, including the multidisciplinary collaboration between specialists (emergency physicians, surgeons, radiologists) and identification of the optimal interventional treatment and post-operative care [[125]]. Traditional circulating biomarkers do not represent a satisfactory and reliable support in the initial patient screening as well, in which, on the contrary, the molecular/genetic evaluation can be diriment. Genetic testing, especially that which interrogates several genes at once in a parallel approach that is, at present, undoubtedly preferable to the cascade one, has long been included in the diagnostic flowchart for TAA diagnosis. First of all, it allows for the identification of a co-existing condition with TAA, such as MFS or LDS, thus directing the most appropriate management in terms of periodic check-ups, time of intervention, risk-recurrence calculation for pregnancies, and screening for first-degree relatives. In addition, the constant implementation of molecular methodologies allowing for the interrogation of the entire genome or transcriptome in an “omic” approach could be, undoubtedly, beneficial for patients’ stratification. In fact, the combination of the data derived from the WES/WGS/RNA-seq approaches can help define profiles that could be highly specific for subgroups of TAA patients, not to mention the potential use of those data in deepening knowledge about the disease’s onset and progression as well as for identifying new targets for therapy. Even with the considerable limitations characterizing the omic approaches (production of a large amount of bioinformatic data that need to be correctly interpreted, safely stored, and validated through functional studies; the possibility of VUSs and incidental findings), the future benefits they may represent for the improvement of the TAA diagnostic work-up have to be considered and perhaps should be addressed more closely and in greater detail in the international guidelines
References
- Bossone, E.; Eagle, K.A. Epidemiology and Management of Aortic Disease: Aortic Aneurysms and Acute Aortic Syndromes. Nat. Rev. Cardiol. 2021, 18, 331–348.
- Kuzmik, G.A.; Sang, A.X.; Elefteriades, J.A. Natural History of Thoracic Aortic Aneurysms. J. Vasc. Surg. 2012, 56, 565–571.
- Clouse, W.D.; Hallett, J.W.; Schaff, H.V.; Spittell, P.C.; Rowland, C.M.; Ilstrup, D.M.; Melton, L.J. Acute Aortic Dissection: Population-Based Incidence Compared with Degenerative Aortic Aneurysm Rupture. Mayo Clin. Proc. 2004, 79, 176–180.
- Nienaber, C.A.; Clough, R.E.; Sakalihasan, N.; Suzuki, T.; Gibbs, R.; Mussa, F.; Jenkins, M.P.; Thompson, M.M.; Evangelista, A.; Yeh, J.S.M.; et al. Aortic Dissection. Nat. Rev. Dis. Primer 2016, 2, 16053.
- Smedberg, C.; Steuer, J.; Leander, K.; Hultgren, R. Sex Differences and Temporal Trends in Aortic Dissection: A Population-Based Study of Incidence, Treatment Strategies, and Outcome in Swedish Patients during 15 Years. Eur. Heart J. 2020, 41, 2430–2438.
- Faggion Vinholo, T.; Zafar, M.A.; Ziganshin, B.A.; Elefteriades, J.A. Nonsyndromic Thoracic Aortic Aneurysms and Dissections—Is Screening Possible? Semin. Thorac. Cardiovasc. Surg. 2019, 31, 628–634.
- Salameh, M.J.; Black, J.H.; Ratchford, E.V. Thoracic Aortic Aneurysm. Vasc. Med. 2018, 23, 573–578.
- Monda, E.; Fusco, A.; Della Corte, A.; Caiazza, M.; Cirillo, A.; Gragnano, F.; Giugliano, M.P.; Citro, R.; Rubino, M.; Esposito, A.; et al. Impact of Regular Physical Activity on Aortic Diameter Progression in Paediatric Patients with Bicuspid Aortic Valve. Pediatr. Cardiol. 2021, 42, 1133–1140.
- Rohde, S.; Zafar, M.A.; Ziganshin, B.A.; Elefteriades, J.A. Thoracic Aortic Aneurysm Gene Dictionary. Asian Cardiovasc. Thorac. Ann. 2021, 29, 682–696.
- Chou, E.L.; Lindsay, M.E. The Genetics of Aortopathies: Hereditary Thoracic Aortic Aneurysms and Dissections. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 136–148.
- Erbel, R.; Aboyans, V.; Boileau, C.; Bossone, E.; Di Bartolomeo, R.; Eggebrecht, H.; Evangelista, A.; Falk, V.; Frank, H.; Gaemperli, O.; et al. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases. Kardiol. Pol. 2014, 72, 1169–1252.
- Harris, S.L.; Lindsay, M.E. Role of Clinical Genetic Testing in the Management of Aortopathies. Curr. Cardiol. Rep. 2021, 23, 10.
- Elefteriades, J.A.; Sang, A.; Kuzmik, G.; Hornick, M. Guilt by Association: Paradigm for Detecting a Silent Killer (Thoracic Aortic Aneurysm). Open Heart 2015, 2, e000169.
- Quintana, R.A.; Taylor, W.R. Cellular Mechanisms of Aortic Aneurysm Formation. Circ. Res. 2019, 124, 607–618.
- Albornoz, G.; Coady, M.A.; Roberts, M.; Davies, R.R.; Tranquilli, M.; Rizzo, J.A.; Elefteriades, J.A. Familial Thoracic Aortic Aneurysms and Dissections—Incidence, Modes of Inheritance, and Phenotypic Patterns. Ann. Thorac. Surg. 2006, 82, 1400–1405.
- Biddinger, A.; Rocklin, M.; Coselli, J.; Milewicz, D.M. Familial Thoracic Aortic Dilatations and Dissections: A Case Control Study. J. Vasc. Surg. 1997, 25, 506–511.
- Coady, M.A.; Davies, R.R.; Roberts, M.; Goldstein, L.J.; Rogalski, M.J.; Rizzo, J.A.; Hammond, G.L.; Kopf, G.S.; Elefteriades, J.A. Familial Patterns of Thoracic Aortic Aneurysms. Arch. Surg. 1999, 134, 361–367.
- Cannon Albright, L.A.; Camp, N.J.; Farnham, J.M.; MacDonald, J.; Abtin, K.; Rowe, K.G. A Genealogical Assessment of Heritable Predisposition to Aneurysms. J. Neurosurg. 2003, 99, 637–643.
- Meester, J.A.N.; Vandeweyer, G.; Pintelon, I.; Lammens, M.; Van Hoorick, L.; De Belder, S.; Waitzman, K.; Young, L.; Markham, L.W.; Vogt, J.; et al. Loss-of-Function Mutations in the X-Linked Biglycan Gene Cause a Severe Syndromic Form of Thoracic Aortic Aneurysms and Dissections. Genet. Med. 2017, 19, 386–395.
- Takeda, N.; Komuro, I. Genetic Basis of Hereditary Thoracic Aortic Aneurysms and Dissections. J. Cardiol. 2019, 74, 136–143.
- Baldwin, A.K.; Simpson, A.; Steer, R.; Cain, S.A.; Kielty, C.M. Elastic Fibres in Health and Disease. Expert Rev. Mol. Med. 2013, 15, e8.
- Szabo, Z. Aortic Aneurysmal Disease and Cutis Laxa Caused by Defects in the Elastin Gene. J. Med. Genet. 2005, 43, 255–258.
- Guemann, A.-S.; Andrieux, J.; Petit, F.; Halimi, E.; Bouquillon, S.; Manouvrier-Hanu, S.; Van De Kamp, J.; Boileau, C.; Hanna, N.; Jondeau, G.; et al. ELN Gene Triplication Responsible for Familial Supravalvular Aortic Aneurysm. Cardiol. Young 2015, 25, 712–717.
- Kent, K.C.; Crenshaw, M.L.; Goh, D.L.M.; Dietz, H.C. Genotype-Phenotype Correlation in Patients with Bicuspid Aortic Valve and Aneurysm. J. Thorac. Cardiovasc. Surg. 2013, 146, 158–165.e1.
- Weerakkody, R.; Ross, D.; Parry, D.A.; Ziganshin, B.; Vandrovcova, J.; Gampawar, P.; Abdullah, A.; Biggs, J.; Dumfarth, J.; Ibrahim, Y.; et al. Targeted Genetic Analysis in a Large Cohort of Familial and Sporadic Cases of Aneurysm or Dissection of the Thoracic Aorta. Genet. Med. Off. J. Am. Coll. Med. Genet. 2018, 20, 1414–1422.
- Ostberg, N.P.; Zafar, M.A.; Ziganshin, B.A.; Elefteriades, J.A. The Genetics of Thoracic Aortic Aneurysms and Dissection: A Clinical Perspective. Biomolecules 2020, 10, 182.
- Franken, R.; Groenink, M.; de Waard, V.; Feenstra, H.M.A.; Scholte, A.J.; van den Berg, M.P.; Pals, G.; Zwinderman, A.H.; Timmermans, J.; Mulder, B.J.M. Genotype Impacts Survival in Marfan Syndrome. Eur. Heart J. 2016, 37, 3285–3290.
- Guo, D.; Regalado, E.S.; Gong, L.; Duan, X.; Santos-Cortez, R.L.P.; Arnaud, P.; Ren, Z.; Cai, B.; Hostetler, E.M.; Moran, R.; et al. LOX Mutations Predispose to Thoracic Aortic Aneurysms and Dissections. Circ. Res. 2016, 118, 928–934.
- Lee, V.S.; Halabi, C.M.; Hoffman, E.P.; Carmichael, N.; Leshchiner, I.; Lian, C.G.; Bierhals, A.J.; Vuzman, D.; Brigham Genomic Medicine; Mecham, R.P.; et al. Loss of Function Mutation in LOX Causes Thoracic Aortic Aneurysm and Dissection in Humans. Proc. Natl. Acad. Sci. USA 2016, 113, 8759–8764.
- Kuang, S.-Q.; Medina-Martinez, O.; Guo, D.-C.; Gong, L.; Regalado, E.S.; Reynolds, C.L.; Boileau, C.; Jondeau, G.; Prakash, S.K.; Kwartler, C.S.; et al. FOXE3 Mutations Predispose to Thoracic Aortic Aneurysms and Dissections. J. Clin. Investig. 2016, 126, 948–961.
- Schubert, J.A.; Landis, B.J.; Shikany, A.R.; Hinton, R.B.; Ware, S.M. Clinically Relevant Variants Identified in Thoracic Aortic Aneurysm Patients by Research Exome Sequencing. Am. J. Med. Genet. A 2016, 170, 1288–1294.
- Milewicz, D.M.; Guo, D.-C.; Tran-Fadulu, V.; Lafont, A.L.; Papke, C.L.; Inamoto, S.; Kwartler, C.S.; Pannu, H. Genetic Basis of Thoracic Aortic Aneurysms and Dissections: Focus on Smooth Muscle Cell Contractile Dysfunction. Annu. Rev. Genom. Hum. Genet. 2008, 9, 283–302.
- Chen, M.H.; Choudhury, S.; Hirata, M.; Khalsa, S.; Chang, B.; Walsh, C.A. Thoracic Aortic Aneurysm in Patients with Loss of Function Filamin A Mutations: Clinical Characterization, Genetics, and Recommendations. Am. J. Med. Genet. A. 2018, 176, 337–350.
- Zhu, L.; Vranckx, R.; Khau Van Kien, P.; Lalande, A.; Boisset, N.; Mathieu, F.; Wegman, M.; Glancy, L.; Gasc, J.-M.; Brunotte, F.; et al. Mutations in Myosin Heavy Chain 11 Cause a Syndrome Associating Thoracic Aortic Aneurysm/Aortic Dissection and Patent Ductus Arteriosus. Nat. Genet. 2006, 38, 343–349.
- Luyckx, I.; Loeys, B.L. Curriculum topic: Disease of the aorta and trauma to the aorta and heart The Genetic Architecture of Non-Syndromic Thoracic Aortic Aneurysm. Heart Br. Card. Soc. 2015, 101, 1678–1684.
- Renard, M.; Francis, C.; Ghosh, R.; Scott, A.F.; Witmer, P.D.; Adès, L.C.; Andelfinger, G.U.; Arnaud, P.; Boileau, C.; Callewaert, B.L.; et al. Clinical Validity of Genes for Heritable Thoracic Aortic Aneurysm and Dissection. J. Am. Coll. Cardiol. 2018, 72, 605–615.
- Ponińska, J.K.; Bilińska, Z.T.; Truszkowska, G.; Michalak, E.; Podgórska, A.; Stępień-Wojno, M.; Chmielewski, P.; Lutyńska, A.; Płoski, R. Good Performance of the Criteria of American College of Medical Genetics and Genomics/Association for Molecular Pathology in Prediction of Pathogenicity of Genetic Variants Causing Thoracic Aortic Aneurysms and Dissections. J. Transl. Med. 2022, 20, 42.
- Quiñones-Pérez, B.; VanNoy, G.E.; Towne, M.C.; Shen, Y.; Singh, M.N.; Agrawal, P.B.; Smith, S.E. Three-Generation Family with Novel Contiguous Gene Deletion on Chromosome 2p22 Associated with Thoracic Aortic Aneurysm Syndrome. Am. J. Med. Genet. A 2018, 176, 560–569.
- MacCarrick, G.; Black, J.H.; Bowdin, S.; El-Hamamsy, I.; Frischmeyer-Guerrerio, P.A.; Guerrerio, A.L.; Sponseller, P.D.; Loeys, B.; Dietz, H.C. Loeys–Dietz Syndrome: A Primer for Diagnosis and Management. Genet. Med. 2014, 16, 576–587.
- Milewicz, D.M.; Regalado, E. Heritable Thoracic Aortic Disease Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993.
- Wooten, E.C.; Iyer, L.K.; Montefusco, M.C.; Hedgepeth, A.K.; Payne, D.D.; Kapur, N.K.; Housman, D.E.; Mendelsohn, M.E.; Huggins, G.S. Application of Gene Network Analysis Techniques Identifies AXIN1/PDIA2 and Endoglin Haplotypes Associated with Bicuspid Aortic Valve. PLoS ONE 2010, 5, e8830.
- Takeda, N.; Morita, H.; Fujita, D.; Inuzuka, R.; Taniguchi, Y.; Imai, Y.; Hirata, Y.; Komuro, I. Congenital Contractural Arachnodactyly Complicated with Aortic Dilatation and Dissection: Case Report and Review of Literature. Am. J. Med. Genet. A 2015, 167, 2382–2387.
- Guo, D.; Gong, L.; Regalado, E.S.; Santos-Cortez, R.L.; Zhao, R.; Cai, B.; Veeraraghavan, S.; Prakash, S.K.; Johnson, R.J.; Muilenburg, A.; et al. MAT2A Mutations Predispose Individuals to Thoracic Aortic Aneurysms. Am. J. Hum. Genet. 2015, 96, 170–177.
- Callewaert, B.; De Paepe, A.; Coucke, P. Arterial Tortuosity Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993.
- Lillie, M.A.; David, G.J.; Gosline, J.M. Mechanical Role of Elastin-Associated Microfibrils in Pig Aortic Elastic Tissue. Connect. Tissue Res. 1998, 37, 121–141.
- Creamer, T.J.; Bramel, E.E.; MacFarlane, E.G. Insights on the Pathogenesis of Aneurysm through the Study of Hereditary Aortopathies. Genes 2021, 12, 183.
- Dietz, H.C.; Cutting, C.R.; Pyeritz, R.E.; Maslen, C.L.; Sakai, L.Y.; Corson, G.M.; Puffenberger, E.G.; Hamosh, A.; Nanthakumar, E.J.; Curristin, S.M.; et al. Marfan Syndrome Caused by a Recurrent de Novo Missense Mutation in the Fibrillin Gene. Nature 1991, 352, 337–339.
- Csiszar, K. Lysyl Oxidases: A Novel Multifunctional Amine Oxidase Family. In Progress in Nucleic Acid Research and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2001; Volume 70l, pp. 1–32. ISBN 978-0-12-540070-1.
- Zentner, D.; James, P.; Bannon, P.; Jeremy, R. Familial Aortopathies—State of the Art Review. Heart Lung Circ. 2020, 29, 607–618.
- Papke, C.L.; Yanagisawa, H. Fibulin-4 and Fibulin-5 in Elastogenesis and beyond: Insights from Mouse and Human Studies. Matrix Biol. 2014, 37, 142–149.
- De Figueiredo Borges, L.; Jaldin, R.G.; Dias, R.R.; Stolf, N.A.G.; Michel, J.-B.; Gutierrez, P.S. Collagen Is Reduced and Disrupted in Human Aneurysms and Dissections of Ascending Aorta. Hum. Pathol. 2008, 39, 437–443.
- Combs, M.D.; Knutsen, R.H.; Broekelmann, T.J.; Toennies, H.M.; Brett, T.J.; Miller, C.A.; Kober, D.L.; Craft, C.S.; Atkinson, J.J.; Shipley, J.M.; et al. Microfibril-Associated Glycoprotein 2 (MAGP2) Loss of Function Has Pleiotropic Effects in Vivo. J. Biol. Chem. 2013, 288, 28869–28880.
- Kolb, M.; Margetts, P.J.; Sime, P.J.; Gauldie, J. Proteoglycans Decorin and Biglycan Differentially Modulate TGF-β-Mediated Fibrotic Responses in the Lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L1327–L1334.
- Guo, D.-C.; Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Yu, R.K.; Avidan, N.; Bourgeois, S.; Estrera, A.L.; Safi, H.J.; Sparks, E.; et al. Mutations in Smooth Muscle α-Actin (ACTA2) Lead to Thoracic Aortic Aneurysms and Dissections. Nat. Genet. 2007, 39, 1488–1493.
- Morisaki, H.; Akutsu, K.; Ogino, H.; Kondo, N.; Yamanaka, I.; Tsutsumi, Y.; Yoshimuta, T.; Okajima, T.; Matsuda, H.; Minatoya, K.; et al. Mutation of ACTA2 Gene as an Important Cause of Familial and Nonfamilial Nonsyndromatic Thoracic Aortic Aneurysm and/or Dissection (TAAD). Hum. Mutat. 2009, 30, 1406–1411.
- Disabella, E.; Grasso, M.; Gambarin, F.I.; Narula, N.; Dore, R.; Favalli, V.; Serio, A.; Antoniazzi, E.; Mosconi, M.; Pasotti, M.; et al. Risk of Dissection in Thoracic Aneurysms Associated with Mutations of Smooth Muscle Alpha-Actin 2 (ACTA2). Heart Br. Card. Soc. 2011, 97, 321–326.
- Lu, H.; Fagnant, P.M.; Bookwalter, C.S.; Joel, P.; Trybus, K.M. Vascular Disease-Causing Mutation R258C in ACTA2 Disrupts Actin Dynamics and Interaction with Myosin. Proc. Natl. Acad. Sci. USA 2015, 112, E4168–E4177.
- Kim, H.; McCulloch, C.A. Filamin A Mediates Interactions between Cytoskeletal Proteins That Control Cell Adhesion. FEBS Lett. 2011, 585, 18–22.
- Siddiqui, S.T.; Fisher, S.D. Heritable FLNA Gene Mutation in a Patient with Thoracic Aortic Aneurysm. JACC Case Rep. 2022, 4, 87–90.
- Pomianowski, P.; Elefteriades, J.A. The Genetics and Genomics of Thoracic Aortic Disease. Ann. Cardiothorac. Surg. 2013, 2, 271–279.
- Daugherty, A.; Chen, Z.; Sawada, H.; Rateri, D.L.; Sheppard, M.B. Transforming Growth Factor-β in Thoracic Aortic Aneurysms: Good, Bad, or Irrelevant? J. Am. Heart Assoc. 2017, 6, e005221.
- Michel, J.-B.; Jondeau, G.; Milewicz, D.M. From Genetics to Response to Injury: Vascular Smooth Muscle Cells in Aneurysms and Dissections of the Ascending Aorta. Cardiovasc. Res. 2018, 114, 578–589.
- MacFarlane, E.G.; Parker, S.J.; Shin, J.Y.; Ziegler, S.G.; Creamer, T.J.; Bagirzadeh, R.; Bedja, D.; Chen, Y.; Calderon, J.F.; Weissler, K.; et al. Lineage-Specific Events Underlie Aortic Root Aneurysm Pathogenesis in Loeys-Dietz Syndrome. J. Clin. Investig. 2019, 129, 659–675.
- Lindsay, M.E.; Dietz, H.C. Lessons on the Pathogenesis of Aneurysm from Heritable Conditions. Nature 2011, 473, 308–316.
- Guo, D.-C.; Regalado, E.S.; Pinard, A.; Chen, J.; Lee, K.; Rigelsky, C.; Zilberberg, L.; Hostetler, E.M.; Aldred, M.; Wallace, S.E.; et al. LTBP3 Pathogenic Variants Predispose Individuals to Thoracic Aortic Aneurysms and Dissections. Am. J. Hum. Genet. 2018, 102, 706–712.
- Schepers, D.; Tortora, G.; Morisaki, H.; MacCarrick, G.; Lindsay, M.; Liang, D.; Mehta, S.G.; Hague, J.; Verhagen, J.; van de Laar, I.; et al. A Mutation Update on the LDS-Associated Genes TGFB2/3 and SMAD2/3. Hum. Mutat. 2018, 39, 621–634.
- Cecconi, M.; Manfrin, M.; Moraca, A.; Zanoli, R.; Colonna, P.L.; Bettuzzi, M.G.; Moretti, S.; Gabrielli, D.; Perna, G.P. Aortic Dimensions in Patients with Bicuspid Aortic Valve without Significant Valve Dysfunction. Am. J. Cardiol. 2005, 95, 292–294.
- Pepe, G.; Nistri, S.; Giusti, B.; Sticchi, E.; Attanasio, M.; Porciani, C.; Abbate, R.; Bonow, R.O.; Yacoub, M.; Gensini, G.F. Identification of Fibrillin 1 Gene Mutations in Patients with Bicuspid Aortic Valve (BAV) without Marfan Syndrome. BMC Med. Genet. 2014, 15, 23.
- Boyum, J.; Fellinger, E.K.; Schmoker, J.D.; Trombley, L.; McPartland, K.; Ittleman, F.P.; Howard, A.B. Matrix Metalloproteinase Activity in Thoracic Aortic Aneurysms Associated with Bicuspid and Tricuspid Aortic Valves. J. Thorac. Cardiovasc. Surg. 2004, 127, 686–691.
- Rabkin, S.W. Differential Expression of MMP-2, MMP-9 and TIMP Proteins in Thoracic Aortic Aneurysm - Comparison with and without Bicuspid Aortic Valve: A Meta-Analysis. Vasa 2014, 43, 433–442.
- Martin-Blazquez, A.; Heredero, A.; Aldamiz-Echevarria, G.; Martin-Lorenzo, M.; Alvarez-Llamas, G. Non-Syndromic Thoracic Aortic Aneurysm: Cellular and Molecular Insights. J. Pathol. 2021, 254, 229–238.
- Geng, L.; Wang, W.; Chen, Y.; Cao, J.; Lu, L.; Chen, Q.; He, R.; Shen, W. Elevation of ADAM10, ADAM17, MMP-2 and MMP-9 Expression with Media Degeneration Features CaCl2-Induced Thoracic Aortic Aneurysm in a Rat Model. Exp. Mol. Pathol. 2010, 89, 72–81.
- Ren, P.; Zhang, L.; Xu, G.; Palmero, L.C.; Albini, P.T.; Coselli, J.S.; Shen, Y.H.; LeMaire, S.A. ADAMTS-1 and ADAMTS-4 Levels Are Elevated in Thoracic Aortic Aneurysms and Dissections. Ann. Thorac. Surg. 2013, 95, 570–577.
- Wilton, E.; Bland, M.; Thompson, M.; Jahangiri, M. Matrix Metalloproteinase Expression in the Ascending Aorta and Aortic Valve. Interact. Cardiovasc. Thorac. Surg. 2008, 7, 37–40.
- Evangelista, A.; Isselbacher, E.M.; Bossone, E.; Gleason, T.G.; Eusanio, M.D.; Sechtem, U.; Ehrlich, M.P.; Trimarchi, S.; Braverman, A.C.; Myrmel, T.; et al. Insights from the International Registry of Acute Aortic Dissection: A 20-Year Experience of Collaborative Clinical Research. Circulation 2018, 137, 1846–1860.
- Silaschi, M.; Byrne, J.; Wendler, O. Aortic Dissection: Medical, Interventional and Surgical Management. Heart 2017, 103, 78–87.
- Hagan, P.G.; Nienaber, C.A.; Isselbacher, E.M.; Bruckman, D.; Karavite, D.J.; Russman, P.L.; Evangelista, A.; Fattori, R.; Suzuki, T.; Oh, J.K.; et al. The International Registry of Acute Aortic Dissection (IRAD): New Insights into an Old Disease. JAMA 2000, 283, 897.
- Vilacosta, I.; San Román, J.A.; di Bartolomeo, R.; Eagle, K.; Estrera, A.L.; Ferrera, C.; Kaji, S.; Nienaber, C.A.; Riambau, V.; Schäfers, H.-J.; et al. Acute Aortic Syndrome Revisited: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 78, 2106–2125.
- White, A.; Broder, J.; Mando-Vandrick, J.; Wendell, J.; Crowe, J. Acute Aortic Emergencies—Part 2 Aortic Dissections. Adv. Emerg. Nurs. J. 2013, 35, 28–52.
- Von Kodolitsch, Y.; Csösz, S.K.; Koschyk, D.H.; Schalwat, I.; Loose, R.; Karck, M.; Dieckmann, C.; Fattori, R.; Haverich, A.; Berger, J.; et al. Intramural Hematoma of the Aorta: Predictors of Progression to Dissection and Rupture. Circulation 2003, 107, 1158–1163.
- Manabe, T.; Imoto, K.; Uchida, K.; Doi, C.; Takanashi, Y. Decreased Tissue Inhibitor of Metalloproteinase-2/Matrix Metalloproteinase Ratio in the Acute Phase of Aortic Dissection. Surg. Today 2004, 34, 220–225.
- Koullias, G.J.; Ravichandran, P.; Korkolis, D.P.; Rimm, D.L.; Elefteriades, J.A. Increased Tissue Microarray Matrix Metalloproteinase Expression Favors Proteolysis in Thoracic Aortic Aneurysms and Dissections. Ann. Thorac. Surg. 2004, 78, 2106–2110.
- Del Porto, F.; di Gioia, C.; Tritapepe, L.; Ferri, L.; Leopizzi, M.; Nofroni, I.; De Santis, V.; Della Rocca, C.; Mitterhofer, A.P.; Bruno, G.; et al. The Multitasking Role of Macrophages in Stanford Type A Acute Aortic Dissection. Cardiology 2014, 127, 123–129.
- Landenhed, M.; Engström, G.; Gottsäter, A.; Caulfield, M.P.; Hedblad, B.; Newton-Cheh, C.; Melander, O.; Smith, J.G. Risk Profiles for Aortic Dissection and Ruptured or Surgically Treated Aneurysms: A Prospective Cohort Study. J. Am. Heart Assoc. 2015, 4, e001513.
- Hahn, A.W.A.; Jonas, U.; Bühler, F.R.; Resink, T.J. Activation of Human Peripheral Monocytes by Angiotensin II. FEBS Lett. 1994, 347, 178–180.
- Stumpf, C.; Jukic, J.; Yilmaz, A.; Raaz, D.; Schmieder, R.E.; Daniel, W.G.; Garlichs, C.D. Elevated VEGF-Plasma Levels in Young Patients with Mild Essential Hypertension. Eur. J. Clin. Investig. 2009, 39, 31–36.
- Derosa, G.; D’Angelo, A.; Ciccarelli, L.; Piccinni, M.N.; Pricolo, F.; Salvadeo, S.; Montagna, L.; Gravina, A.; Ferrari, I.; Galli, S.; et al. Matrix Metalloproteinase-2, -9, and Tissue Inhibitor of Metalloproteinase-1 in Patients with Hypertension. Endothel. J. Endothel. Cell Res. 2006, 13, 227–231.
- Elefteriades, J.A. Natural History of Thoracic Aortic Aneurysms: Indications for Surgery, and Surgical versus Nonsurgical Risks. Ann. Thorac. Surg. 2002, 74, S1877–S1880, discussion S1892–S1898.
- Pape, L.A.; Tsai, T.T.; Isselbacher, E.M.; Oh, J.K.; O’gara, P.T.; Evangelista, A.; Fattori, R.; Meinhardt, G.; Trimarchi, S.; Bossone, E.; et al. Aortic Diameter > or =5.5 Cm Is Not a Good Predictor of Type A Aortic Dissection: Observations from the International Registry of Acute Aortic Dissection (IRAD). Circulation 2007, 116, 1120–1127.
- Davies, R.R.; Goldstein, L.J.; Coady, M.A.; Tittle, S.L.; Rizzo, J.A.; Kopf, G.S.; Elefteriades, J.A. Yearly Rupture or Dissection Rates for Thoracic Aortic Aneurysms: Simple Prediction Based on Size. Ann. Thorac. Surg. 2002, 73, 17–27, discussion 27–28.
- Gawinecka, J.; Schönrath, F.; von Eckardstein, A. Acute Aortic Dissection: Pathogenesis, Risk Factors and Diagnosis. Swiss Med. Wkly. 2017, 147, w14489.
- Pyeritz, R.E. Recent Progress in Understanding the Natural and Clinical Histories of the Marfan Syndrome. Trends Cardiovasc. Med. 2016, 26, 423–428.
- Weinsaft, J.W.; Devereux, R.B.; Preiss, L.R.; Feher, A.; Roman, M.J.; Basson, C.T.; Geevarghese, A.; Ravekes, W.; Dietz, H.C.; Holmes, K.; et al. Aortic Dissection in Patients with Genetically Mediated Aneurysms. J. Am. Coll. Cardiol. 2016, 67, 2744–2754.
- Den Hartog, A.W.; Franken, R.; Zwinderman, A.H.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; de Waard, V.; Pals, G.; Mulder, B.J.M.; Groenink, M. The Risk for Type B Aortic Dissection in Marfan Syndrome. J. Am. Coll. Cardiol. 2015, 65, 246–254.
- Pape, L.A.; Awais, M.; Woznicki, E.M.; Suzuki, T.; Trimarchi, S.; Evangelista, A.; Myrmel, T.; Larsen, M.; Harris, K.M.; Greason, K.; et al. Presentation, Diagnosis, and Outcomes of Acute Aortic Dissection: 17-Year Trends from the International Registry of Acute Aortic Dissection. J. Am. Coll. Cardiol. 2015, 66, 350–358.
- Tran-Fadulu, V.; Pannu, H.; Kim, D.H.; Vick, G.W.; Lonsford, C.M.; Lafont, A.L.; Boccalandro, C.; Smart, S.; Peterson, K.L.; Hain, J.Z.; et al. Analysis of Multigenerational Families with Thoracic Aortic Aneurysms and Dissections Due to TGFBR1 or TGFBR2 Mutations. J. Med. Genet. 2009, 46, 607–613.
- Jondeau, G.; Ropers, J.; Regalado, E.; Braverman, A.; Evangelista, A.; Teixedo, G.; De Backer, J.; Muiño-Mosquera, L.; Naudion, S.; Zordan, C.; et al. International Registry of Patients Carrying TGFBR1 or TGFBR2 Mutations: Results of the MAC (Montalcino Aortic Consortium). Circ. Cardiovasc. Genet. 2016, 9, 548–558.
- Van de Laar, I.M.B.H.; Oldenburg, R.A.; Pals, G.; Roos-Hesselink, J.W.; de Graaf, B.M.; Verhagen, J.M.A.; Hoedemaekers, Y.M.; Willemsen, R.; Severijnen, L.-A.; Venselaar, H.; et al. Mutations in SMAD3 Cause a Syndromic Form of Aortic Aneurysms and Dissections with Early-Onset Osteoarthritis. Nat. Genet. 2011, 43, 121–126.
- Laterza, D.; Ritelli, M.; Zini, A.; Colombi, M.; Dell’Acqua, M.L.; Vandelli, L.; Bigliardi, G.; Verganti, L.; Vallone, S.; Vincenzi, C.; et al. Novel Pathogenic TGFBR1 and SMAD3 Variants Identified after Cerebrovascular Events in Adult Patients with Loeys-Dietz Syndrome. Eur. J. Med. Genet. 2019, 62, 103727.
- Zhao, H.; Yang, Y.; Pan, X.; Li, W.; Sun, L.; Guo, J. Identification of Clinically Relevant Variants by Whole Exome Sequencing in Chinese Patients with Sporadic Non-Syndromic Type A Aortic Dissection. Clin. Chim. Acta 2020, 506, 160–165.
- Hostetler, E.M.; Regalado, E.S.; Guo, D.-C.; Hanna, N.; Arnaud, P.; Muiño-Mosquera, L.; Callewaert, B.L.; Lee, K.; Leal, S.M.; Wallace, S.E.; et al. SMAD3 Pathogenic Variants: Risk for Thoracic Aortic Disease and Associated Complications from the Montalcino Aortic Consortium. J. Med. Genet. 2019, 56, 252–260.
- Hilhorst-Hofstee, Y.; Scholte, A.J.H.A.; Rijlaarsdam, M.E.B.; van Haeringen, A.; Kroft, L.J.; Reijnierse, M.; Ruivenkamp, C.A.L.; Versteegh, M.I.M.; Pals, G.; Breuning, M.H. An Unanticipated Copy Number Variant of Chromosome 15 Disrupting SMAD3 Reveals a Three-Generation Family at Serious Risk for Aortic Dissection. Clin. Genet. 2013, 83, 337–344.
- Lee, S.-T.; Kim, J.-A.; Jang, S.-Y.; Kim, D.-K.; Kim, J.-W.; Ki, C.-S. A Novel COL3A1 Gene Mutation in Patient with Aortic Dissected Aneurysm and Cervical Artery Dissections. Heart Vessels 2008, 23, 144–148.
- Koitabashi, N.; Yamaguchi, T.; Fukui, D.; Nakano, T.; Umeyama, A.; Toda, K.; Funada, R.; Ishikawa, M.; Kawamura, R.; Okada, K.; et al. Peripartum Iliac Arterial Aneurysm and Rupture in a Patient with Vascular Ehlers-Danlos Syndrome Diagnosed by Next-Generation Sequencing. Int. Heart J. 2018, 59, 1180–1185.
- Makrygiannis, G.; Loeys, B.; Defraigne, J.-O.; Sakalihasan, N. Cervical Artery Dissections and Type A Aortic Dissection in a Family with a Novel Missense COL3A1 Mutation of Vascular Type Ehlers–Danlos Syndrome. Eur. J. Med. Genet. 2015, 58, 634–636.
- Nakamura, M.; Yajima, J.; Oikawa, Y.; Ogasawara, K.; Uejima, T.; Abe, K.; Aizawa, T. Vascular Ehlers-Danlos Syndrome. J. Cardiol. 2009, 53, 458–462.
- Shields, L.B.E.; Rolf, C.M.; Davis, G.J.; Hunsaker III, J.C. Sudden and Unexpected Death in Three Cases of Ehlers-Danlos Syndrome Type IV*: SUDDEN DEATH IN EHLERS-DANLOS TYPE IV. J. Forensic Sci. 2010, 55, 1641–1645.
- Schwarze, U.; Goldstein, J.A.; Byers, P.H. Splicing Defects in the COL3A1 Gene: Marked Preference for 5′ (Donor) Spice-Site Mutations in Patients with Exon-Skipping Mutations and Ehlers-Danlos Syndrome Type IV. Am. J. Hum. Genet. 1997, 61, 1276–1286.
- Sakai, K.; Toda, M.; Kyoyama, H.; Nishimura, H.; Kojima, A.; Kuwabara, Y.; Kobayashi, Y.; Kikuchi, S.; Hirata, Y.; Moriyama, G.; et al. Vascular Ehlers-Danlos Syndrome with a Novel Missense Mutation in COL3A1: A Man in His 50s with Aortic Dissection after Interventional Treatment for Hemothorax as the First Manifestation. Intern. Med. Tokyo Jpn. 2019, 58, 3441–3447.
- Shalhub, S.; Black, J.H.; Cecchi, A.C.; Xu, Z.; Griswold, B.F.; Safi, H.J.; Milewicz, D.M.; McDonnell, N.B. Molecular Diagnosis in Vascular Ehlers-Danlos Syndrome Predicts Pattern of Arterial Involvement and Outcomes. J. Vasc. Surg. 2014, 60, 160–169.
- Meienberg, J.; Rohrbach, M.; Neuenschwander, S.; Spanaus, K.; Giunta, C.; Alonso, S.; Arnold, E.; Henggeler, C.; Regenass, S.; Patrignani, A.; et al. Hemizygous Deletion of COL3A1, COL5A2, and MSTN Causes a Complex Phenotype with Aortic Dissection: A Lesson for and from True Haploinsufficiency. Eur. J. Hum. Genet. 2010, 18, 1315–1321.
- Wang, Z.; Zhuang, X.; Chen, B.; Wen, J.; Peng, F.; Liu, X.; Wei, M. 99-Case Study of Sporadic Aortic Dissection by Whole Exome Sequencing Indicated Novel Disease-Associated Genes and Variants in Chinese Population. BioMed Res. Int. 2020, 2020, 7857043.
- Chen, Y.; Sun, Y.; Li, Z.; Li, C.; Xiao, L.; Dai, J.; Li, S.; Liu, H.; Hu, D.; Wu, D.; et al. Identification of COL3A1 Variants Associated with Sporadic Thoracic Aortic Dissection: A Case-Control Study. Front. Med. 2021, 15, 438–447.
- Frank, M.; Albuisson, J.; Ranque, B.; Golmard, L.; Mazzella, J.-M.; Bal-Theoleyre, L.; Fauret, A.-L.; Mirault, T.; Denarié, N.; Mousseaux, E.; et al. The Type of Variants at the COL3A1 Gene Associates with the Phenotype and Severity of Vascular Ehlers–Danlos Syndrome. Eur. J. Hum. Genet. 2015, 23, 1657–1664.
- Pepin, M.G.; Schwarze, U.; Rice, K.M.; Liu, M.; Leistritz, D.; Byers, P.H. Survival Is Affected by Mutation Type and Molecular Mechanism in Vascular Ehlers-Danlos Syndrome (EDS Type IV). Genet. Med. Off. J. Am. Coll. Med. Genet. 2014, 16, 881–888.
- Saratzis, A.; Bown, M.J. The Genetic Basis for Aortic Aneurysmal Disease. Heart Br. Card. Soc. 2014, 100, 916–922.
- Guo, D.; Regalado, E.; Casteel, D.E.; Santos-Cortez, R.L.; Gong, L.; Kim, J.J.; Dyack, S.; Horne, S.G.; Chang, G.; Jondeau, G.; et al. Recurrent Gain-of-Function Mutation in PRKG1 Causes Thoracic Aortic Aneurysms and Acute Aortic Dissections. Am. J. Hum. Genet. 2013, 93, 398–404.
- Teodoro Jerves; Andrea Beaton; Paul Kruszka; The genetic workup for structural congenital heart disease. American Journal of Medical Genetics Part C: Seminars in Medical Genetics 2019, 184, 178-186, 10.1002/ajmg.c.31759.
- Christina M. Rigelsky; Rocio T. Moran; Genetics of syndromic and nonsyndromic aortopathies. Current Opinion in Pediatrics 2019, 31, 694-701, 10.1097/mop.0000000000000836.
- Jj Gómez de Diego; Comments on the 2014 ESC Guidelines on the Diagnosis and Treatment of Aortic Diseases. Revista Española de Cardiología (English Edition) 2015, 68, 179-184, 10.1016/j.rec.2014.12.003.
- John Augoustides; Michael Andritsos; Hiratzka Lf; Bakris Gl; Beckman Ja; Bersin Rm; Carr Vf; Eagle Ka; Hermann Lk; Isselbacher Em; et al.Kazerooni EaKouchoukos NtLytle BwMilewicz DmReich DlSen SShinn JaSvensson LgWilliams DmAmerican College of Cardiology Foundation/American Heart Association Task Force on Practice GuidelinesAmerican Association for Thoracic SurgeryAmerican College of RadiologyAmerican Stroke AssociationSociety Of Cardiovascular AnesthesiologistsSociety For Cardiovascular Angiography And InterventionsSociety Of Interventional RadiologySociety Of Thoracic SurgeonsSociety for Vascular Medicine 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine.. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2010, 121, e266–e369, 10.3410/f.2671959.2334057.
- Dong-Chuan Guo; Ellen M. Hostetler; Yuxin Fan; Richard J. Kulmacz; Di Zhang; Deborah A. Nickerson; Suzanne M. Leal; Scott A. LeMaire; Ellen S. Regalado; Dianna M. Milewicz; et al. Heritable Thoracic Aortic Disease Genes in Sporadic Aortic Dissection. Journal of the American College of Cardiology 2017, 70, 2728-2730, 10.1016/j.jacc.2017.09.1094.
- Kelli L. Hicks; Peter H. Byers; Elina Quiroga; Melanie G. Pepin; Sherene Shalhub; Testing patterns for genetically triggered aortic and arterial aneurysms and dissections at an academic center. Journal of Vascular Surgery 2018, 68, 701-711, 10.1016/j.jvs.2017.12.023.
- Tim Ripperger; Hans Dieter Tröger; Jörg Schmidtke; The genetic message of a sudden, unexpected death due to thoracic aortic dissection. Forensic Science International 2009, 187, 1-5, 10.1016/j.forsciint.2009.01.020.
- Christopher K. Mehta; Andre Y. Son; Matthew C. Chia; Ashley N. Budd; Bradley D. Allen; Patricia Vassallo; Andrew W. Hoel; William J. Brady; Jose V. Nable; Management of acute aortic syndromes from initial presentation to definitive treatment. The American Journal of Emergency Medicine 2021, 51, 108-113, 10.1016/j.ajem.2021.10.016.