Definition: Alzheimer’s disease (AD) is an age-related dementia and neurodegenerative disorder, characterized by Aβ and tau protein deposition impairing learning, memory, and suppressing synaptic plasticity of neurons. Increasing evidence suggests that there is a link between the glucose and glutamate alterations with age that down-regulates glucose utilization reducing glutamate levels in AD patients. Deviations in brain energy metabolism reinforce the development of AD by hampering glutamate levels in the brain. Glutamate is a nonessential amino acid and the major excitatory neurotransmitter synthesized from glucose. Alterations in cerebral glucose and glutamate levels precede the deposition of Aβ plaques. In the brain, over 40% of neuronal synapses are glutamatergic and disturbances in glutamatergic function have been implicated in the pathophysiology of AD. Nevertheless, targeting the glutamatergic system seems to be a promising strategy to develop novel, improved therapeutics for AD. Thus, the present review will extensively cover recent findings on the dysregulation of glutamatergic signaling in AD and will highlight the molecular mechanisms through which the modulation of glutamatergic receptors and transporters might exert beneficial effects in AD treatment.
- Alzheimer's disease
- Glutamate
- NMDA
- EAAT
- Long-term potentiation
- Long-term depression
- AMPA
- amyloid-β
- tau
- amyloid precursor protein
- ionotropic receptors
- metabotropic receptors
Note:All the information in this draft can be edited by authors. And the entry will be online only after authors edit and submit it.
1. Introduction
Alzheimer’s disease (AD) is the most common and prevalent neurodegenerative disease with memory dysfunction and cognitive impairment, affecting nearly 46.8 million people worldwide, as reported by the World Health Organisation. AD is characterized by extracellular deposition of amyloid-β (Aβ) senile plaques and intracellular accumulation of neurofibrillary tangles (NFTs) [1]. Aβ is a short peptide that is produced from amyloid precursor protein (APP), a type I integral membrane glycoprotein which undergoes cleavage by both amyloidogenic and non-amyloidogenic pathways [2]. Amyloidogenic pathway produces soluble APPβ (sAPPβ) fragment, Aβ, and APP intracellular domain (AICD) through sequential cleavage of APP by β-secretase and γ-secretase; non-amyloidogenic pathway generates soluble APPα (sAPPα), P3 peptide and AICD by α-secretase and γ-secretase [3]. Out of all Aβ species, soluble oligomeric Aβ1-42 is considered as the most neurotoxic product obtained after cleavage of APP [4]. Aβ was first identified in the extracellular component and later studies confirmed its presence in neuronal intracellular regions such as the endosome [5], endoplasmic reticulum [6], and trans-Golgi network [7]. Aβ in mitochondria reacts with proteins impairing oxidative phosphorylation and increasing reactive oxygen species (ROS) that damage the neuronal membrane [8][9][10][8,9,10].
Aβ peptides are involved in cognitive dysfunction by inducing synaptotoxicity through Aβ aggregation. Aβ species can be found at the synapse as low-molecular-weight (LMW) dimers, trimers, and tetramers) and high-molecular-weight (HMW) oligomers [11]. HMW Aβ oligomers have greater binding affinity in hippocampal neuronal synapses when compared to LMW Aβ oligomers [12]. However, LMW Aβ oligomers induce more cognitive dysfunction and HMW Aβ oligomers cause a transient decrease in cognitive functions [13]. Nevertheless, HMW oligomers can dissociate into LMW species impairing synaptic functions [14] and in contrast, dissociation of Aβ oligomers into monomers in vivo could reduce Aβ pathology and synaptotoxicity [15]. Aβ oligomers target the excitatory synapses with greater affinity and alter the structure and functions of the synapses [16].
Tau is a microtubule-associated protein (MAP) engaged in the stability of microtubules in neurons and also regulates synaptic function [17]. Hyperphosphorylation of tau results in the formation of NFTs, which is a major pathological hallmark of AD. Tau phosphorylation acts as an indicator of aberrant activities of kinases and phosphatases in AD [18]. In dendrites, hyperphosphorylation of tau forms fibrils, which appear as neuropil threads and as NFTs in axon and the somatodendritic section of neurons [19][20][19,20]. Tau protein undergoes phosphorylation by proline-directed protein kinases (PDPKs) or non-proline-directed protein kinases (non-PDPKs) [21]. PDPKs include Cyclin-Dependent Kinase-5 (Cdk-5), Glycogen synthase kinase-3 (GSK-3), and extracellular signal-related protein kinase (ERK). The irregular activity of Cdk-5 in AD causes tau hyperphosphorylation, loss of dendritic spines, and deterioration of synaptic plasticity [22]. Non-PDPKs comprise of protein kinase A (PKA), Casein kinase 1 (CK1), and Casein kinase 2 (CK2) [18]. Among the kinases identified to be responsible for tau hyperphosphorylation, GSK-3β plays an important role in the pathological changes of tau protein in AD [23]. Data collected in humans show increased activation of GSK-3β in early-stage AD [24], while a consistent inhibition was observed in late-stage AD [25][26][25,26], thus suggesting that GSK-3β-mediated tau phosphorylation is among the earliest events during the progression of the pathology. Increasing evidence shows that tau in dendrites plays a key role in Aβ-induced detrimental effects. Of note, tau knockout mice showed a decrease in extrasynaptic N-methyl-d-aspartate (NMDA) receptor activity in the hippocampus contributing to neuroprotective effects [27]. Accumulation of Aβ plaques in synapse and the infiltration of tau into dendritic spines reduce excitatory glutamatergic synaptic transmission leading to cognitive impairments [28].
In the central nervous system (CNS), glutamate is the primary excitatory neurotransmitter acting on both ionotropic and metabotropic receptors. It is at the crossroad between multiple metabolic pathways and plays an important role in the functions of learning and memory. The activity of glutamatergic neurons is compromised in AD due to the destruction of synapse and neuronal death and its deficit can influence memory, cognition, and behavior, including cortical and hippocampal processing [29][30][29,30]. On the other hand, the pathological accumulation of glutamate can induce neurotoxicity due to time-related exposure, over-stimulating the post-synaptic response causing an increase in the entry of Ca2+ into neurons [31]. Since currently available therapies of AD include cholinesterase inhibitors, NMDA partial antagonist memantine, more information about AD-related changes in glutamate neurotransmission would be highly relevant to better understand its mechanisms of action and to optimize the treatment [32][33][32,33]. In fact, treatment using memantine has improved cognition, behavior, global function but the degree of efficacy remains to be fully determined.
2. Glucose Levels Affect Glutamate Content in AD
AD patients commonly suffer from other co-morbidities (stress, depression, diabetes, renal disease, etc.) that increase the complexity of AD pathogenesis [34][35][36][37][38][39][40][34,35,36,37,38,39,40]. Among all the co-morbidities, diabetes remains the most prevailing and significant risk in developing AD [41][42][43][44][45][46][41,42,43,44,45,46]. Familial AD patients exhibit alterations in the cerebral glucose metabolism before the manifestation of amyloid plaques that might be the causative reason for AD development [47][48][47,48].
Glucose is the main energy substrate for the brain cells (glial cells and neurons). It is now well established that aberrations in cerebral glucose utilization, glycolysis, and oxidative metabolism are associated with cognitive dysfunction in AD [43][48][49][50][51][43,48,49,50,51]. There is a particular pattern of regional brain glucose hypometabolism in AD affecting the parietal and temporal cortices, where the glucose deficit is on the order of 20–25% compared to age-matched, cognitively normal controls [52]. Many studies indicate that presymptomatic brain glucose hypometabolism can be present long before the threshold of cognitive symptoms of AD and could therefore potentially be contributing to the development and/or progression of both the cognitive decline and the neuropathological hallmarks associated with AD. To this regard, although a neuropathological link has not yet been fully demonstrated in humans, increasing evidence in transgenic mouse models of AD suggests that various experimental approaches to diminishing brain glucose supply all drive Aβ overproduction [53]. Moreover, both type-2 diabetes and its associated brain insulin resistance were proposed to favour tau hyper-phosphorylation in AD, although the molecular mechanisms are still unclear [51][54][55][51,54,55].
During the course of time, glucose alterations in the brain have a detrimental effect on the glutamatergic system due to an imbalance in glutamate availability. Neurons and glial cells work in tight cooperation in the glutamate/glutamine cycle and this cycle is connected with the energy metabolism, and, in turn, with the availability of glucose to the brain. Glutamate can be converted into α-ketoglutarate and vice-versa and serves as a substrate in the tricarboxylic acid (TCA) cycle. Research on APPswe/PSEN1dE9 mice showed that prior to amyloid plaque deposition, brain energy metabolism is affected leading to changes in glucose metabolism, reduced TCA cycle activity, decreased adenosine triphosphate (ATP) synthesis in isolated mitochondria were observed demonstrating that alterations in cerebral energy metabolism precede the amyloid plaques formation [56]. The conversion of glutamate to glutamine by the enzyme glutamine synthetase requires ATP and it has been estimated that 70% of the signaling energy comes from the oxidation of glucose [57]. Therefore, impairment in glucose metabolism impacts the glutamate receptor-mediated signal pathway leading to initial stages of memory impairment observed in patients affected by AD [58][59][58,59]. Figure 1 describes how alterations in cerebral glucose levels affect glutamate output leading to neuronal death in AD.
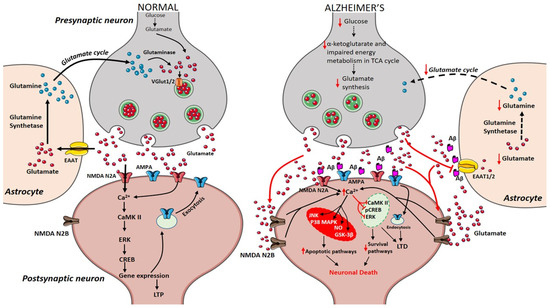
Figure 1. Presynaptic terminals release glutamate activating ionotropic receptors on postsynaptic neurons. In normal conditions, N-methyl-d-aspartate (NMDA) NR2A activation induces an increase in calcium levels favoring the induction of long-term potentiation (LTP) through metabolic pathways (extracellular signal-related protein kinase (ERK), CaMK II, cyclic adenosine monophosphate response element-binding protein (CREB)). Excess glutamate left is taken up by astrocytes through EAAT2 converting into glutamine and glutamate by glutamine synthetase and glutaminase respectively (black arrows). The synthesized glutamate is transported into vesicles by VGlut1/2. Conversely in Alzheimer’s disease (AD), Aβ oligomers interfere with NMDA receptors increasing the spillover of glutamate (red arrows) to extrasynaptic sites activating NMDA NR2B receptors increasing excess calcium levels inhibiting prosurvival pathways. Imbalance in the glutamate/glutamine cycle (black-dashed arrows) is also reported in the figure.
3. Therapeutics for AD
Drug discovery, research, and development for AD are strenuous and challenging. Over hundreds of drugs have failed and, currently, there are 132 agents in clinical trials for AD treatment. Since 2003, no new drugs and no disease-modifying treatments (DMTs) have been approved for AD [60][209]. Current treatment of AD includes drugs targeting the cholinergic system such as donepezil, rivastigmine, galantamine; drug acting on the glutamatergic system like memantine, and drugs that intervene both cholinergic and glutamatergic system namely Namzaric, a combination of memantine and donepezil [61][210]. Compounds acting on glutamatergic receptors, transporters, and their key findings are listed in table 1.
3.1. Modulators of Ionotropic Receptors
The non-competitive NMDA receptor antagonist memantine has an inhibitory effect on NMDA receptor-mediated excitotoxicity, improves cognition, slowdowns disease progression, and reduces tau phosphorylation [32][33][62][63][32,33,211,226]. Phencyclidine, ketamine, and MK-801 (dizocilpine) target NMDA receptors but their clinical applications are hampered due to severe side effects [64][214]. Memantine inhibits neuronal excitotoxicity by inhibiting extrasynaptic Ca2+ influx, improving symptoms in patients with moderate to severe AD [65][227]. However, memantine preferentially targets HMW Aβ-induced synaptotoxicity rescuing from both neuronal oxidative stress and transient memory impairment but unable to prevent LMW Aβ-induced persistent cognitive deficit [13]. An improved NMDA receptor antagonist nitromemantine protects neuronal synapses both in vitro and in vivo by selectively blocking the aberrant extrasynaptic activity over physiological synaptic NMDA receptor activity [66][124].
Uncaria rhynchophylla is a medicinal herb that contains oxindole alkaloid, rhynchophylline that restores LTP and alleviates Aβ-induced activation of extrasynaptic NMDA receptors. Rhynchophylline is a significant active compound that protects from deficits in spatial learning and memory induced by soluble Aβ oligomers. Moreover, rhynchophylline also prevents the hyperactivation of extrasynaptic NMDA receptors by reducing postsynaptic currents in AD mice [67][216]. Another medicinal plant Pulsatilla Chinensis contains a natural triterpenoid saponin compound anemoside A3 (AA3) that modulates synaptic connectivity and memory enhancement. AA3 increases serine phosphorylation of AMPA receptors subunit of GluA1 that is required for AMPA receptors trafficking at synapses. In the hippocampus, AA3 increases the expression of monoamine neurotransmitters and neurotrophin, a brain-derived neurotrophic factor. Furthermore, AA3 acts as a non-competitive NMDA receptor modulator protective against overexcitation and ischemic brain injury [68][217].
3.2. Modulators of mGlu Receptors
3.2.1. mGlu5 Receptor Modulators
mGlu5 receptor is coupled with heterotrimeric G protein Gαq/11 and its activation results in the release of intracellular Ca2+ that is linked to numerous neurodegenerative diseases [69][167]. Genetic deletion of mGlu5 receptor rescues cognitive decline and AD pathogenesis in APPswe/PS1EΔ9 AD mouse model [70][181]. Besides, selective blockade of mGlu5 receptor activity with a negative allosteric modulator (NAM) 2-chloro-4-[2-[2,5-dimethyl-1-[4-(trifluoromethoxy)phe-nyl]imidazol-4-yl]ethynyl] pyridine (CTEP) improved cognition in AD mice [71][218]. CTEP rescues cognitive functions by decreasing Aβ levels through ZBTB16-mediated autophagy activation in APPswe/PS1EΔ9 mice [72][228]. However, the efficacy of CTEP remains inconsistent with disease progression. In 15-month-old APPswe/PS1EΔ9 mice, loss of CTEP efficacy is found after 36 weeks of treatment due to the abolished contribution of ZBTB16 and mammalian target of rapamycin (mTOR)-mediated signaling. This data suggests that the pathological role of mGlu5 receptors may shift during the course of disease progression and proper therapeutic strategies should be amended for beneficial outcomes [73][229].
Furthermore, the selective blockade of mGlu5 receptors with 3-((2-methyl-1,3-thiazol-4-yl)ethynyl)pyridine (MTEP) reversed the learning and memory deficits in AD mice by rescuing synaptic dysfunction [70][74][181,186]. A key finding suggests that the silent allosteric modulator (SAM) BMS-984923 was able to rescue the established memory deficits in AD mice with normal mGlu5 receptor signaling in APPswe/PS1EΔ9 mouse model of AD. BMS-984923 is able to potentially inhibit mGlu5-PrPc interactions preventing Aβ-induced pathological signaling [75][219]. Moreover, mGlu5 receptors are also involved in regulating the release of inflammatory factors and ATP in non-neuronal cells such as microglia and astrocytes [76][230]. Non-selective group I/II mGlu receptor antagonist LY341495, reported to improve synaptic plasticity and blocking Aβ-enhanced long-term depression [77][224]. Moreover, pretreatment with mGlu1/5 receptor agonist, 3,5-dihydroxyphenylglycone (DHPG) also decreased Aβ-enhanced LTD [78][220]. mGlu5 receptor non-competitive antagonist SIB1757, prevented Aβ-induced reduction of NMDA receptors when neurons were pretreated with this molecule [79][173].
3.2.2. mGlu2/3 Receptor Agonist/Antagonists
mGlu2/3 receptor antagonists demonstrated pro-cognitive effects in the Morris water maze test [80][199], novel recognition test [81][231], and social recognition test [82][232]. Orthosteric mGlu2/3 receptor agonists like LY379268 exerts mGlu3 receptor signaling, protecting neurons through the production of TGF-β1 and glial cell line-derived neurotrophic factors (GDNF) [83][84][85][86][193,221,222,223]. Durand et al. (2017) showed that apart from mGlu3 receptor agonists, protective effects through astrocyte-derived neutrophins, neuronal mGlu3 receptor activation also protects against Aβ-induced toxicity that disagrees with a previous report by Caraci et al., 2011. LY379268 injection upregulates brain-derived neurotrophic factor (BDNF) mRNA and protein levels in neurons of the cerebral cortex and hippocampus [87][233], group II mGlu receptor agonist (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG-IV) increased BDNF mRNA only in microglial cells [88][234], while another study found an increase in BDNF mRNA levels after treating cultured astrocytes with LY379268 [89][197]. mGlu2 receptor positive allosteric modulator (PAM), N-4’-cyano-biphenyl-3-yl)-N-(3 pyridinylmethyl)-ethanesulfonamide hydrochloride (LY566332) has increased Aβ-induced neurodegeneration, while this effect is prevented by treatment with mGlu2/3 receptor agonist (2S,1′S,2′S)-2-(9-xanthylmethyl)-2-(2′-carboxycyclopropyl) glycine (LY341495) [83][193].
3.3. EAAT2 Activators
EAAT2 impairment is implicated in excess glutamate accumulation at the synaptic cleft leading to neurodegeneration in AD. The compounds that increase the activity of EAAT2 may have therapeutic benefits and neuroprotection. To investigate the cognitive benefit of restored EAAT2 in APPSw,Ind mice, a novel translational activator (LDN/OSU-0212320) is used as a pharmacological approach that restored EAAT2 protein significantly with improved cognitive functions and reduced Aβ plaques [90][69]. In a virtual screening approach study, two molecules GT949 and GT951 were identified as PAM of EAAT2 in cultured cells that enhanced glutamate uptake showing neuroprotective properties [91][225].
Table 1. List of compounds acting on glutamatergic receptors, transporters, and their key findings.
|
Receptors |
Drugs |
Mechanisms |
Key Findings |
Preclinical and Clinical Studies/Approval Status |
References |
|
NMDA |
Memantine |
Non-competitive NMDAR antagonist |
Improves cognition Slow-down disease progression Reduce tau phosphorylation |
Approved by EMEA in 2002 and by USFDA in 2003 |
|
|
|
Phencyclidine |
NMDAR antagonist |
Psychotomimetic |
No clinical applications |
[64][214] |
|
|
Ketamine |
Non-competitive NMDAR antagonist |
Psychotomimetic |
No clinical applications |
[64][214] |
|
|
MK-801 (Dizocilpine) |
Non-competitive NMDA blocker |
Cardiovascular side effects |
No clinical applications |
[64][214] |
|
|
Nitromemantine |
Selective inhibition of extrasynaptic NMDAR |
Ameliorates Aβ-induced synaptic loss |
In vivo studies in α7nAChR-knockout, hAPP-J20 Tg, and 3× Tg AD mice |
[94][106] |
|
|
Rhynchophylline (oxindole alkaloid) |
Prevented excessive activation of Aβ1-42-induced postsynaptic extrasynaptic NMDARs |
Rescues Aβ1–42-induced spatial dysfunction and LTP impairment. Limited application due to low water solubility, low concentration in brain tissue and low bioavailability |
In vivo studies in Adult male Sprague-Dawley rats and C57BL/6 mice are under research to improve brain targeted delivery |
|
|
AMPA |
Anemoside A3 (triterpenoid saponin) |
Modulates AMPA receptor |
Improves memory and synaptic strength |
In vivo studies in C57BL/6 (C57) mice |
[68][217] |
|
mGluR5 |
2-chloro-4-[2-[2,5-dimethyl-1-[4-(trifluoromethoxy)phe-nyl]imidazol-4-yl]ethynyl] pyridine (CTEP) |
Negative Allosteric Modulator |
Improves cognition |
In vivo studies and CTEP is analogue of phase II molecule Basimglurant (RO4917523) |
[71][218] |
|
|
3-((2-methyl-1,3-thiazol- 4-yl)ethynyl)pyridine (MTEP) |
mGluR5-specific antagonist |
Rescues from synaptic dysfunction and ameliorates learning and memory |
In vivo studies in APPswe/PS1ΔE9 mice, APP/PS1 transgenic mice |
|
|
|
BMS-984923 |
Silent Allosteric modulator- |
Rescues memory deficits and prevents Aβ-induced pathological signaling |
In vivo studies in APPswe/PS1DE9 (APP/PS1) transgenic model mice In vitro studies in HEK293T cells |
[75][219] |
|
|
LY341495 |
Non-selective group I/II mGluR antagonist |
Completely blocks Aβ-induced LTD |
In vivo studies in male Wistar rats |
[78][220] |
|
|
3,5-dihydroxyphenylglycone (DHPG) |
Group 1 mGluR (mGluR1/5) agonist |
Prevents Aβ-induced LTD |
In vivo studies in male Wistar rats |
[98][220] |
|
|
SIB1757-[6-methyl-2-(phenylazo)-3-pyridinol] |
Noncompetitive mGluR5 antagonist |
Prevents Aβ-induced reduction of NMDARs |
In vivo studies in mGluR5 knockout mice |
[99][155] |
|
mGluR2/3 |
LY379268 |
Orthosteric mGluR2/3 agonists |
Protects neurons through TGF-β and GDNF production |
In Mixed Cultures of Mouse Cortical Cells; mGlu2 and mGlu3 receptor knockout mice |
|
|
|
LY341495 |
mGluR2/3 antagonists |
Blocks release of Aβ42 |
In cultured astrocytes and cultured neurons lacking mGlu2 receptors; TgCRND8 mice overexpressing a mutant human APP 695 |
|
|
EAAT2 |
LDN/OSU-0212320 |
EAAT2 translational activator |
Improves cognitive functions |
In vivo studies in APPSw,Ind mice |
[90][69] |
|
|
GT949, GT951 |
Positive allosteric modulators |
Enhances glutamate transport |
In vivo studies in C57BL/6 mice |
[91][225] |
4. Conclusions
Neurodegeneration as in AD is a multifactorial disease, which is why a general pathological mechanism and appropriate treatment have not been found, but various etiological hypotheses have been proposed in order to understand a little about the etiology of AD. To this regard, glutamate seems to play major roles in part because of its abundance in brain tissue and in part because it is at the crossroad of multiple metabolic pathways. In fact, glutamate is the major mediator of excitatory signals in SNC, and both too much glutamate and too little glutamate are harmful. When this delicate balance is disrupted, the perturbations of glutamate neurotransmission have severe consequences, leading to the onset of AD. Although it has a pivotal role in the etiology of AD, the glutamatergic system offers many pharmacological tools and therapeutical targets in order to slow down the disease.
In this regard, antagonists of the NMDA receptor damper the excitotoxicity induced by glutamate in AD. Memantine and its combination with donepezil are approved by the Food and Drug Administration to treat moderate to severe AD. However, these current medications are not able to completely rescue the brain cells from the damage of AD progression. Thus, there is a lot of need to focus on and develop some disease-modifying drugs that could slow the progression of AD. Some medicinal herbs contain active components, like rhynchophylline, and AA3, which restore LTP by inhibiting the activation of extrasynaptic NMDA receptor and enhance cognition by acting on AMPA receptors respectively. Moreover, mGlu2/3 ligands could be used as antagonists for AD treatment, and mGlu5 receptor modulators rescue from cognitive decline. Of note, PAMs of EAAT2 could block glutamate-mediated excitotoxicity by increasing the glutamate clearance. Thus, it is clear that the future to treat AD is through a multi-drug and multi-model approach using combinations of potential drugs for the treatment. The lack of successful drug developments in AD has provided the opportunity to develop agents that could modify AD progression. Aiming at the glutamatergic system is one such target that could be beneficial in the treatment of AD by reducing glutamate levels rescuing from glutamate-induced excitotoxicity.