Aging is a process associated with the detriment of normal physiological functions, which leads to the manifestation of diverse diseases such as cardiovascular and neurodegenerative diseases, joint degenerative diseases, and metabolic diseases such as diabetes, among others. Lysosomes are heterogeneous organelles enclosed by a lipid bilayer and filled with hydrolytic enzymes. The lysosomes are traditionally described as the subcellular structures where the degradation of other organelles and macromolecules takes place, a fundamental process for maintaining cellular proteostasis. There are several degradation processes in which the lysosomes are involved. If the substrate reaching the lysosomes comes from the extracellular environment, the degradation process is called endocytosis. If the material to be digested comes from the cell itself, the process is classified as autophagy. The lysosomes are also involved in plasma membrane repair through a mechanism called lysosomal exocytosis. During aging, damage in cellular organelles disbalances the cellular homeostatic processes. Lysosomal dysfunction is emerging as an important factor that could regulate the production of inflammatory molecules, metabolic cellular state, or mitochondrial function. Thus, lysosomal alkalinization, amino acid storage, iron disturbances and lipofuscin accummulation are characteristic features of the lysosome during aging.
- lysosome
- aging
- senescence
- mTORC
- lipofuscin
- TFEB
- inflammation
1. Lysosomes
1.1. Lysosomal Structure and Components
To understand the lysosomal participation in these processes, the composition and structure of the organelle must be understood. The lysosome is delimited by a bilipidic membrane that encloses an acid lumen, where the degradative reactions take place. This lumen contains approximately 60 hydrolases that are active at acidic pH and participate in multistep catabolic processes to orchestrate the degradation of organelles and macromolecules to monomers [1]. Lysosomes have a role in protein degradation, since they contain proteases such as cathepsins. However, they are also involved in the degradation of other macromolecules such as lipids, which are catabolized by lipases such as the lysosomal acid lipase LIPA [2]. Nucleases such as RNase T2 or DNase II participate in nucleic acid degradation [3], and enzymes such as the acid alpha-glucosidase are important for polysaccharide degradation [4]. Macromolecules catabolized into monomers inside the lysosomes can be reused in anaplerotic reactions, connecting lysosomal function with cellular metabolism. The lysosomal membrane is covered in its internal part with a large glycocalyx, a thick layer of polysaccharides that prevent lysosomal membrane self-digestion, avoiding acid content leakage into the cytosol [1]. The lysosomal membrane contains key proteins for the fusion of the lysosome with autophagosomes, endosomes, or with the plasma membrane as well as pumps and channels that are fundamental for the transport of ions and molecules and to maintain lysosomal acidification [5]. Lysosomal acidification is tightly controlled. These organelles are acidified mainly by the action of the v-ATPase, which hydrolyses ATP to pump protons into the lysosomal lumen. This process generates a transmembrane voltage that is dissipated by the action of ion channels [5]. The correct functioning of these ion channels is important not just to dissipate the electrogenic gradient, but also for v-ATPase to continue introducing protons against the gradient. Indeed, an increase in lysosomal pH can have detrimental consequences on the digestion carried out by lysosomal hydrolases, since these enzymes are active at low pH (pH 4–5).1.2. Lysosomal Biogenetic Pathways and Metabolic Integration
Lysosomal biogenesis is important for lysosomal adaptation to different situations and to replace disrupted or dysfunctional lysosomes. Similarly, under nutrient deprivation conditions, when autophagy is induced, lysosomal biogenesis is activated since the number of lysosomes must increase. Additionally, when a cell replicates, lysosomes have to be produced to be dispersed between the daughter cells. Diverse mechanisms participating in lysosome biogenesis have been described [6]. The mammalian target of rapamycin (mTORC) is a protein kinase involved in the regulation of cell growth and metabolism. mTORC is the main component of mTORC1 and mTORC2, two protein complexes that are distinguished by their accessory proteins. Aside from mTORC, mTORC1 contains the regulatory-associated protein of mTOR (RAPTOR), and mTORC2 is characterized by the presence of the rapamycin-insensitive subunit companion of mTOR (RICTOR) [7]. One mechanism that regulates lysosomal biogenesis depends on mTORC1 (mammalian target of rapamycin complex 1) activity, which converges in the activation or inhibition of TFEB (Figure 1). mTORC1 can sense the metabolic state of the cell. In nutrient-rich conditions, active mTORC1 binds with TFEB, the master transcriptional factor in the regulation of lysosome biogenesis and function, at the lysosomal surface [8]. mTORC1 phosphorylates TFEB, causing its binding, sequestration, and consequent inactivation by the regulatory 14-3-3 proteins in the cytosol. When the cell requires nutrients, mTORC1 is inactivated and TFEB translocates to the nucleus where it promotes the transcription of its target genes, thus enhancing lysosome biogenesis and autophagy [6]. This set of genes, involved in lysosome biogenesis, is known as the CLEAR (Coordinated Lysosomal Expression and Regulation) gene network. TFEB is the main regulator of this network, but other transcription factors from the MiT/TFE family to which TFEB belongs such as TFE3, MITF, and TFEC can also regulate the transcription of these genes [9]. TFEB also regulates the expression of v-ATPase subunits, so mTORC1 indirectly regulates v-ATPase activity and the acidification process [10]. The v-ATPase subunit association is also a tightly regulated process. Although this mechanism has been better studied in yeast, it is known that in mammals, the assembly of v-ATPase is promoted by amino acid starvation and by glucose starvation [11]. Furthermore, TFEB enhances the expression of genes related to lipid catabolism as a response to cell starvation [12]. TFEB also enhances the transcription of PGC1-α, which is the master regulator of mitochondrial biogenesis and participates in mitochondrial clearance [13].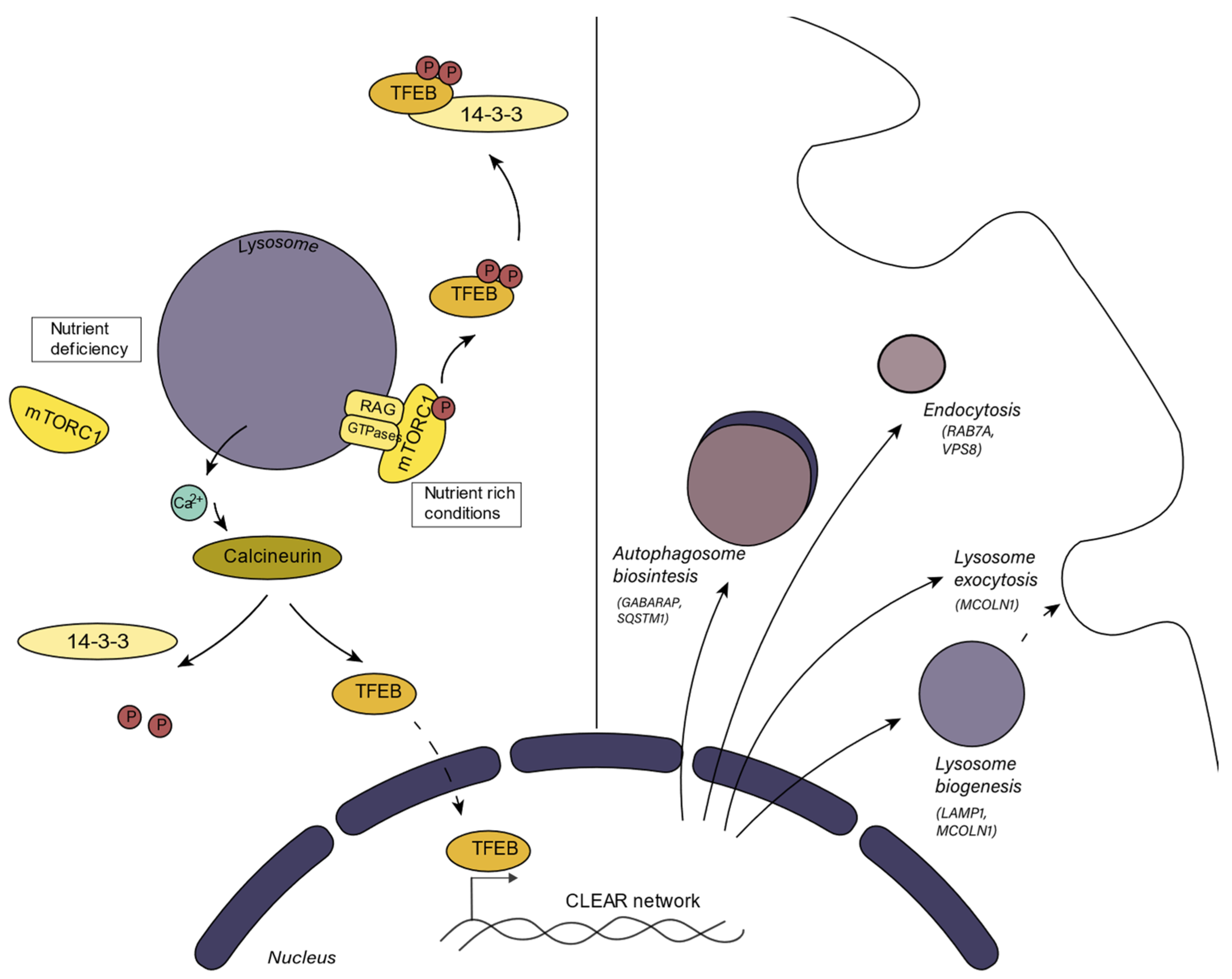
2. Processes in which the Lysosome Participates
2.1. Endocytosis
Endocytosis is a cellular process in which cellular membrane engulfing allows for the internalization of external material. There are several types of endocytosis: phagocytosis, macropinocytosis, clathrin-mediated endocytosis, caveolin-mediated endocytosis, and clathrin-and caveolin-independent endocytosis [15]. In each type of endocytosis, different membrane receptors participate in the engulfing process. After plasma membrane engulfment, an endosome is formed. The newly formed endosome matures to an early endosome that mediates receptor recycling to the membrane. The transition to late endosome is coordinated by small GTPases following a maturation pathway. This maturation allows for the progressive acidification of the endosome, which finally fuses with the lysosome, where it reaches the most acidic pH [16].2.2. Autophagy
Autophagy is a cellular catabolic process that is lysosomal-dependent, in which the cell degrades its own material. It can be classified into three types: microautophagy, chaperone-mediated autophagy, and macroautophagy [17]. Macroautophagy is characterized by the formation of double-membrane vesicles, called autophagosomes, which sequester the substrates to be degraded. It is the most studied process, and it is the only way of eliminating whole organelles, so it is crucial for the removal of damaged organelles.2.3. Mitophagy and Mitochondrial Dysfunction
Lysosomes and mitochondria are tightly interdependent [18]. Several mechanisms have been proposed to explain how lysosomal function interferes with mitochondrial function during aging. Mitophagy has emerged as the main link between these organelles. Mitochondria are double-membrane-bound organelles that play a major role in cellular metabolism since aerobic respiration takes place in these organelles. The TCA cycle and fatty acid oxidation generate reducing agents that donate electrons to the mitochondrial electron transport chain for ATP production. Aside from their function in bioenergetics, mitochondria are organelles that influence cell fate since many enzymes that regulate apoptosis such as BCL-2 proteins or cytochrome c are found in these cellular compartments [19]. When the mitochondria are damaged and the mitochondrial membrane potential decreases, the respiratory chain does not work efficiently and ROS are produced. ROS generate oxidized proteins and lipids that further exacerbate mitochondrial dysfunction. These impaired mitochondria have to be removed, and the main mechanism to eliminate whole organelles is macroautophagy. The selective degradation of deficient mitochondria through macroautophagy is called mitophagy [19]. Thus, lysosomes are directly involved in the maintenance of mechanisms of mitochondria quality control.2.4. Lysosomal Exocytosis
Lysosomes can fuse with the plasma membrane and secrete lysosomal content through exocytosis. This process was first believed to be reserved for specialized cells such as macrophages or melanocytes [20][21], but it has been observed that it occurs in all cells as a repair mechanism of the plasma membrane [22]. Most lysosomes are localized in the perinuclear area, but when the plasma membrane is damaged, lysosomes migrate to the cell periphery to fuse with the plasma membrane promoting the recovery of the membrane. The lysosomal fusion with the membrane is regulated by lysosomal calcium efflux through TRPML1. Precisely, lysosomal exocytosis is frequently studied by the presence of lysosomal proteins such as LAMP1 or TRPML1 in the plasma membrane [23]. Lysosomal exocytosis is a process that also allows to get rid of unprocessed materials and it has been described in the case of lysosomal enzymes. Cathepsin D has been found in extracellular media upon lysosomal alkalinization [24]. Other groups have reported that lysosomes that fuse with the plasma membrane can get rid of autophagosomes [25]. LC3 lipidation is important for lysosomal exocytosis, so its regulation must be closely linked to autophagy [25].3. Lysosomal Age-Related Dysfunctions
Lysosomal function is very important for cellular homeostasis, not only because of the recycling functions it exerts on damaged molecules and dysfunctional organelles, but also because of the impact it has on metabolic pathways or on organelles such as the mitochondria. Most studies are not focused on lysosomal function, but instead cover fields in which the lysosome is involved such as autophagy or mTORC1 status. However, some studies have reported dysfunctions associated with the lysosome in the context of aging (Figure 2).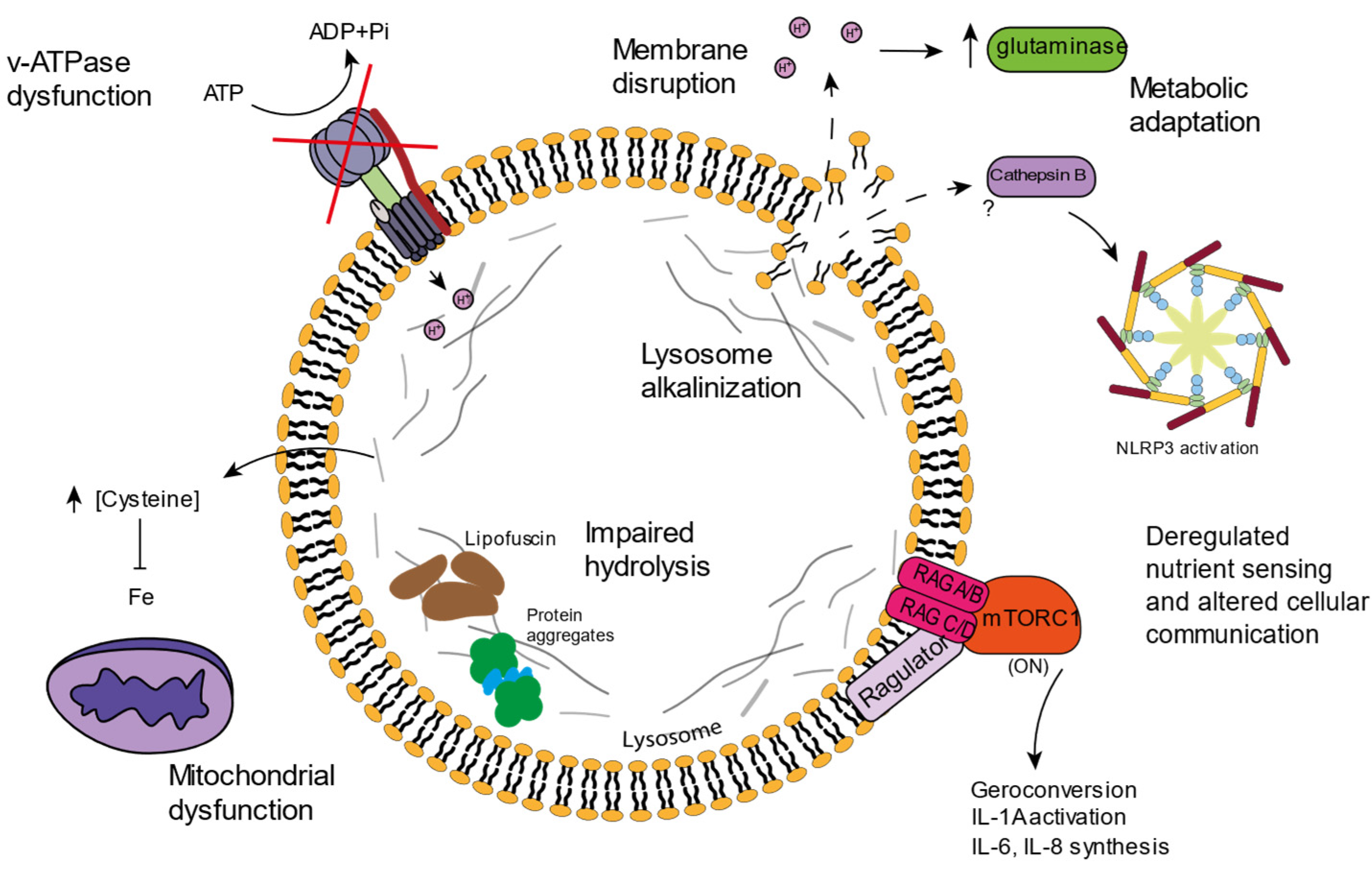
43.1. v-ATPAse Dysfunction and Lysosomal Alkalinization
Although some alterations have been described in luminal enzymes, the lysosomal membrane also has its relevance when it comes to aging. The correct v-ATPase function and lumen acidification has been correlated with delayed aging in yeast [28][29]. Many mutations in the v-ATPase subunits have been associated with neurodegenerative disorders, and animal models carrying mutations in v-ATPase subunits show lysosomal acidification problems accompanied by accelerated aging [30]. Cells lacking v-ATPase subunits also display mitochondrial dysfunctions, highlighting the important relation between lysosomes and mitochondria [31][32].
43.2. Lysosomal Amino Acid Storage and Ion Homeostasis
Lysosomal dysfunctions lead to the accumulation of metabolites, and lysosomal storage diseases are an example of how the incorrect function of one lysosomal enzyme leads to storage problems, which conclude in signs and symptoms at the systemic level [33].
43.3. Lipofuscin
During aging, autophagy and subsequent lysosomal degradation of lipofuscin is impaired, which promotes further aggregation of these granules. Additionally, not only do impaired lysosomes contribute to lipofuscin accumulation during senescence, but the lack of cell division impedes the distribution of these aggregates between the daughter cells, as would occur in proliferative cells. Moreover, the increase in ROS production and lipofuscin accumulation promotes mitochondrial dysfunction, which further exacerbates lysosomal impairment in a positive feedback mechanism [44]. Additionally, lipofuscin accumulation enhances the activity of caspase-3 and promotes lysosomal membrane disruption, having been linked to NLRP3 inflammasome activation and necroptosis induction [45][46].
43.4. Inflammation and Cell Death
Senescent cells show increased mitochondrial mass, and these mitochondria are often dysfunctional. Typically, the mitochondria of senescent cells display low membrane potential, lower mitochondrial ATP production, and increased ROS production [19]. The decreased levels of mitophagy observed in many senescent cells supports a possible mechanism that explains the increase in dysfunctional mitochondria. Lysosomal dysfunction impedes the correct degradation of mitochondria, exacerbating mitochondrial ROS production which, in turn, increases lysosomal damage in a feedback fashion. Both mitochondria and lysosomes have been correlated to SASP production. In fact, mtDNA depletion seems to induce MiDAS with a characteristic SASP pattern that lacks the IL-1 inflammatory arm [5], while lysosomal disruption seems to induce IL-1 activation, enhancing NLRP3 inflammasome function [47]. Lysosomal membrane permeabilization is not only involved in the activation of pro-inflammatory pathways, but it has been suggested to be involved in the mechanisms of cell death, and recently, it has also been associated with pathophysiological conditions [48]. Indeed, some scholars have proposed that lysosomal membrane permeabilization in association with lysosomal quality control mechanisms can determine cell fate, since lysosomal components that are released into the cytosol can trigger the activation of diverse cellular pathways [49]. Several studies have suggested that lysosomal damage is related to apoptosis, necroptosis, and ferroptosis. Apoptosis, the main type of programmed cell death, which is characterized by cytochrome c release from mitochondria, can be fostered by the release of lysosomal cathepsins, which can degrade Bcl-2 [50][51], one of the proteins involved in mitochondrial membrane stability during apoptosis. Similarly, cathepsin D release has been associated to the activation of RIPK1 during necroptosis [52][53], a type of cell death though to be non-programmed, but now known to be regulated by receptor-interacting protein kinases (RIPK). Ferroptosis is a regulated cell death type in which intracellular iron incites the formation of ROS. Lysosomes are closely related to ferroptosis, since these organelles constitute one of the main iron storage places. Lysosomal membrane disruption seems to foster the activation of this cell death pathway [54].54. mTORC and Senescence
mTORC1 seems to be activated during aging and in some senescent cells [55][56]. This activation is evidenced by the phosphorylation of mTORC1 downstream targets. It has been reported that in replicative senescent fibroblasts, p70S6K undergoes phosphorylation by the activity of mTORC1 [57]. Moreover, it has been found that constitutive mTORC1 activation induces premature senescence in fibroblasts carrying tuberous sclerosis complex (TCS) mutations [58]. S6 kinases, phosphorylated and activated by mTORC1, are observed in aged muscle [59] and in the brains of Alzheimer’s disease patients [60]. mTORC1 seems to be fundamental for the processing and activation of some SASP factors such as IL-6, IL-8, or IL-1A, which are, in turn, fundamental players in the inflammation induction that accompanies aging (Figure 2). Rapamycin treatment, which inhibits mTORC1, diminishes the pro-inflammatory phenotype of senescent cells through a reduction in the IL-1A and IL-6 levels [61]. Senescence induction seems to increase mTORC1 activity, which could be related to SASP induction through the TOR-Autophagy Spatial Coupling Compartment (TASCC). It has been suggested that mTORC1 localizes at this TASCC compartment in the Trans-Golgi and favors the synthesis of IL-6 and IL-8 cytokines [62]. Precisely, lysosomes originate from the Trans-Golgi network and mTORC1 activation occurs on the lysosomal membrane. Further studies are necessary to elucidate the relevance of the role of the mTORC1-lysosome on inflammation induction, but several scholars have pointed out the role of lysosomes on inflammasome activation [33][46]. The mTOR pathway can positively or negatively regulate p53, being cell type and stress dependent [63]. In fact, mTORC1 and mTORC2 were reported to participate in the stabilization of cell cycle inhibitors (Figure 3). In MEFS, mTORC1 activation promotes the association of p53 mRNA with ribosomes, leading to p53 translation [64]. The overexpression of the microRNA miR-107 increases MTORC1 activity through PTEN inhibition, resulting in the activation of p16 [65]. Precisely, the loss of PTEN enhances p53 translation, triggered by mTORC1 [66]. Moreover, it has been reported that in PTEN-depleted cells, mTORC1 and mTORC2 bind and phosphorylate p53 at Ser15 [67]. This exhibits the participation of mTORC1 in senescent state regulation.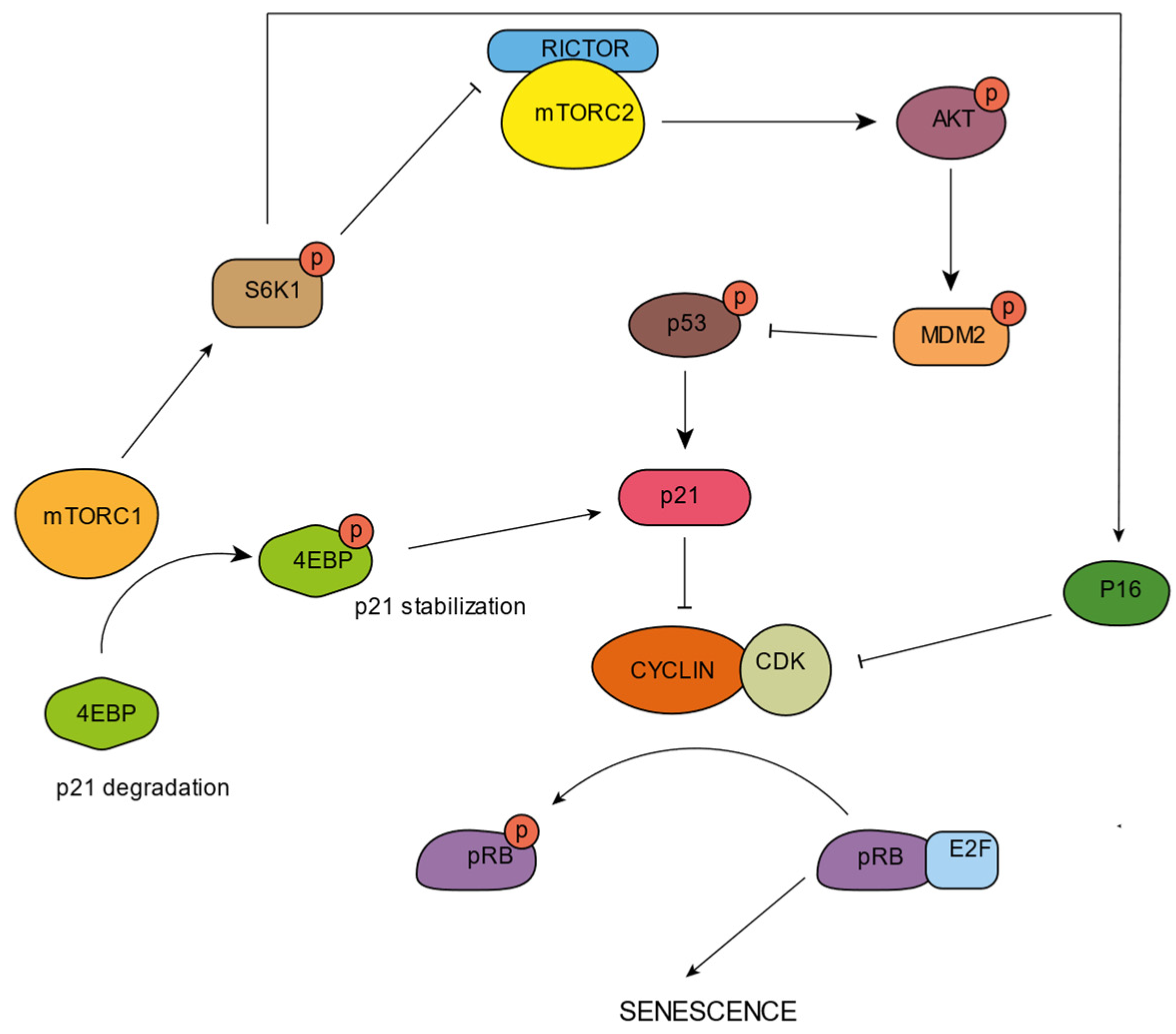
65. Lysosomal Opportunities for Intervention in Aging
References
- Carmine Settembre; Alessandro Fraldi; Diego L. Medina; Andrea Ballabio; Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nature Reviews Molecular Cell Biology 2013, 14, 283-296, 10.1038/nrm3565.
- Ashley M. Thelen; Roberto Zoncu; Emerging Roles for the Lysosome in Lipid Metabolism. Trends in Cell Biology 2017, 27, 833-850, 10.1016/j.tcb.2017.07.006.
- Yuuki Fujiwara; Keiji Wada; Tomohiro Kabuta; Lysosomal degradation of intracellular nucleic acids—multiple autophagic pathways. Journal of Biochemistry 2016, 161, 145-154, 10.1093/jb/mvw085.
- Arnold E. Stütz; Tanja M. Wrodnigg; Carbohydrate-Processing Enzymes of the Lysosome. null 2016, 73, 225-302, 10.1016/bs.accb.2016.08.002.
- Joseph A. Mindell; Lysosomal Acidification Mechanisms. Annual Review of Physiology 2012, 74, 69-86, 10.1146/annurev-physiol-012110-142317.
- Chonglin Yang; Xiaochen Wang; Lysosome biogenesis: Regulation and functions. Journal of Cell Biology 2021, 220, 6, 10.1083/jcb.202102001.
- Mathieu Laplante; David M. Sabatini; mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274-293, 10.1016/j.cell.2012.03.017.
- Carmine Settembre; Roberto Zoncu; Diego L Medina; Francesco Vetrini; Serkan Erdin; SerpilUckac Erdin; Tuong Huynh; Mathieu Ferron; Gerard Karsenty; Michel C Vellard; et al.Valeria FacchinettiDavid M SabatiniAndrea Ballabio A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. The EMBO Journal 2012, 31, 1095-1108, 10.1038/emboj.2012.32.
- Marco Sardiello; Michela Palmieri; Alberto di Ronza; Diego Luis Medina; Marta Valenza; Vincenzo Alessandro Gennarino; Chiara Di Malta; Francesca Donaudy; Valerio Embrione; Roman S. Polishchuk; et al.Sandro BanfiGiancarlo ParentiElena CattaneoAndrea Ballabio A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473-477, 10.1126/science.1174447.
- Samuel Peña-Llopis; Silvia Vega-Rubin-De-Celis; Jacob C Schwartz; Nicholas C Wolff; Tram Anh T Tran; Lihua Zou; Xian-Jin Xie; David R Corey; James Brugarolas; Regulation of TFEB and V-ATPases by mTORC1. The EMBO Journal 2011, 30, 3242-3258, 10.1038/emboj.2011.257.
- Chen-Song Zhang; Bin Jiang; Mengqi Li; Mingjiang Zhu; Yongying Peng; Ya-Lin Zhang; Yu-Qing Wu; Terytty Yang Li; Yu Liang; Zailian Lu; et al.Guili LianQing LiuHuiling GuoZhenyu YinZhiyun YeJiahuai HanJia-Wei WuHuiyong YinShu-Yong LinSheng-Cai Lin The Lysosomal v-ATPase-Ragulator Complex Is a Common Activator for AMPK and mTORC1, Acting as a Switch between Catabolism and Anabolism. Cell Metabolism 2014, 20, 526-540, 10.1016/j.cmet.2014.06.014.
- Carmine Settembre; Rossella De Cegli; Gelsomina Mansueto; Pradip K. Saha; Francesco Vetrini; Orane Visvikis; Tuong Huynh; Annamaria Carissimo; Donna Palmer; Tiemo Jürgen Klisch; et al.Amanda C. WollenbergDiego Di BernardoLawrence ChanJavier E. IrazoquiAndrea Ballabio TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nature 2013, 15, 647-658, 10.1038/ncb2718.
- Heidi Martini-Stoica; Yin Xu; Andrea Ballabio; Hui Zheng; The Autophagy–Lysosomal Pathway in Neurodegeneration: A TFEB Perspective. Trends in Neurosciences 2016, 39, 221-234, 10.1016/j.tins.2016.02.002.
- Yang Li; Meng Xu; Xiao Ding; Chen Yan; Zhiqin Song; Lianwan Chen; Xiahe Huang; Xin Wang; Youli Jian; Guihua Tang; et al.Changyong TangYingtong DiShuzhen MuXuezhao LiuKai LiuTing LiYingchun WangLong MiaoWeixiang GuoXiaojiang HaoChonglin Yang Protein kinase C controls lysosome biogenesis independently of mTORC1. Nature Cell Biology 2016, 18, 1065-1077, 10.1038/ncb3407.
- Gary J. Doherty; Harvey T. McMahon; Mechanisms of Endocytosis. Annual Review of Biochemistry 2009, 78, 857-902, 10.1146/annurev.biochem.78.081307.110540.
- Dmitry Poteryaev; Sunando Datta; Karin Ackema; Marino Zerial; Anne Spang; Identification of the Switch in Early-to-Late Endosome Transition. Cell 2010, 141, 497-508, 10.1016/j.cell.2010.03.011.
- Katherine R. Parzych; Daniel J. Klionsky; An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxidants & Redox Signaling 2014, 20, 460-473, 10.1089/ars.2013.5371.
- Gonzalo Soto-Heredero; Francesc Baixauli; María Mittelbrunn; Interorganelle Communication between Mitochondria and the Endolysosomal System. Frontiers in Cell and Developmental Biology 2017, 5, 95, 10.3389/fcell.2017.00095.
- Guo Chen; Guido Kroemer; Oliver Kepp; Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Frontiers in Cell and Developmental Biology 2020, 8, 200, 10.3389/fcell.2020.00200.
- Carl J. DeSelm; Brian C. Miller; Wei Zou; Wandy L. Beatty; Eline van Meel; Yoshifumi Takahata; Judith Klumperman; Sharon A. Tooze; Steven L. Teitelbaum; Herbert W. Virgin; et al. Autophagy Proteins Regulate the Secretory Component of Osteoclastic Bone Resorption. Developmental Cell 2011, 21, 966-974, 10.1016/j.devcel.2011.08.016.
- Anand K. Ganesan; Hsiang Ho; Brian Bodemann; Sean Petersen; Jayavani Aruri; Shiney Koshy; Zachary Richardson; Lu Q. Le; Tatiana Krasieva; Michael G. Roth; et al.Pat FarmerMichael A. White Genome-Wide siRNA-Based Functional Genomics of Pigmentation Identifies Novel Genes and Pathways That Impact Melanogenesis in Human Cells. PLOS Genetics 2008, 4, e1000298, 10.1371/journal.pgen.1000298.
- Matthias Corrotte; Thiago Castro-Gomes; Lysosomes and plasma membrane repair. null 2019, 84, 1-16, 10.1016/bs.ctm.2019.08.001.
- Brunella Tancini; Sandra Buratta; Federica Delo; Krizia Sagini; Elisabetta Chiaradia; Roberto Maria Pellegrino; Carla Emiliani; Lorena Urbanelli; Lysosomal Exocytosis: The Extracellular Role of an Intracellular Organelle. Membranes 2020, 10, 406, 10.3390/membranes10120406.
- Benny Zhitomirsky; Yehuda G. Assaraf; Lysosomal accumulation of anticancer drugs triggers lysosomal exocytosis. Oncotarget 2017, 8, 45117-45132, 10.18632/oncotarget.15155.
- Sandra Buratta; Brunella Tancini; Krizia Sagini; Federica Delo; Elisabetta Chiaradia; Lorena Urbanelli; Carla Emiliani; Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular. International Journal of Molecular Sciences 2020, 21, 2576, 10.3390/ijms21072576.
- Michael Beck; The Link Between Lysosomal Storage Disorders and More Common Diseases. Journal of Inborn Errors of Metabolism and Screening 2016, 4, 1-8, 10.1177/2326409816682767.
- Ana Toledano-Zaragoza; María Dolores Ledesma; Addressing neurodegeneration in lysosomal storage disorders: Advances in Niemann Pick diseases. Neuropharmacology 2019, 171, 107851, 10.1016/j.neuropharm.2019.107851.
- Adam L. Hughes; Daniel E. Gottschling; An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature 2012, 492, 261-265, 10.1038/nature11654.
- Tobias Wilms; Erwin Swinnen; Elja Eskes; Laura Dolz-Edo; Alice Uwineza; Ruben Van Essche; Joëlle Rosseels; Piotr Zabrocki; Elisabetta Cameroni; Vanessa Franssens; et al.Claudio De VirgilioGertien J. SmitsJoris Winderickx The yeast protein kinase Sch9 adjusts V-ATPase assembly/disassembly to control pH homeostasis and longevity in response to glucose availability. PLOS Genetics 2017, 13, e1006835, 10.1371/journal.pgen.1006835.
- Daniel J. Colacurcio; Ralph A. Nixon; Disorders of lysosomal acidification—The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Research Reviews 2016, 32, 75-88, 10.1016/j.arr.2016.05.004.
- Nicola Baker; Graham Hamilton; Jonathan M. Wilkes; Sebastian Hutchinson; Michael P. Barrett; David Horn; Vacuolar ATPase depletion affects mitochondrial ATPase function, kinetoplast dependency, and drug sensitivity in trypanosomes. Proceedings of the National Academy of Sciences 2015, 112, 9112-9117, 10.1073/pnas.1505411112.
- King Faisal Yambire; Christine Rostosky; Takashi Watanabe; David Pacheu-Grau; Sylvia Torres-Odio; Angela Sanchez-Guerrero; Ola Senderovich; Esther G Meyron-Holtz; Ira Milosevic; Jens Frahm; et al.A. Phillip WestNuno Raimundo Impaired lysosomal acidification triggers iron deficiency and inflammation in vivo. eLife 2019, 8, e51031, 10.7554/elife.51031.
- Calogera M. Simonaro; Lysosomes, Lysosomal Storage Diseases, and Inflammation. Journal of Inborn Errors of Metabolism and Screening 2016, 4, 1-8, 10.1177/2326409816650465.
- Francesca Rizzollo; Sanket More; Peter Vangheluwe; Patrizia Agostinis; The lysosome as a master regulator of iron metabolism. Trends in Biochemical Sciences 2021, 46, 960-975, 10.1016/j.tibs.2021.07.003.
- Bibbin T. Paul; David H. Manz; Frank M. Torti; Suzy V. Torti; Mitochondria and Iron: current questions. Expert Review of Hematology 2016, 10, 65-79, 10.1080/17474086.2016.1268047.
- Rola S. Zeidan; Sung Min Han; Christiaan Leeuwenburgh; Rui Xiao; Iron homeostasis and organismal aging. Ageing Research Reviews 2021, 72, 101510, 10.1016/j.arr.2021.101510.
- Børge G. Nordestgaard; Aram S. Adourian; Jacob J. Freiberg; Yu Guo; Pieter Muntendam; Erling Falk; Risk Factors for Near-Term Myocardial Infarction in Apparently Healthy Men and Women. Clinical Chemistry 2010, 56, 559-567, 10.1373/clinchem.2009.139964.
- Shannon L. Rhodes; Beate Ritz; Genetics of iron regulation and the possible role of iron in Parkinson's disease. Neurobiology of Disease 2008, 32, 183-195, 10.1016/j.nbd.2008.07.001.
- Scott Ayton; Peng Lei; Nigral Iron Elevation Is an Invariable Feature of Parkinson’s Disease and Is a Sufficient Cause of Neurodegeneration. BioMed Research International 2014, 2014, 1-9, 10.1155/2014/581256.
- Jun-Lin Liu; Yong-Gang Fan; Zheng-Sheng Yang; Zhan-You Wang; Chuang Guo; Iron and Alzheimer’s Disease: From Pathogenesis to Therapeutic Implications. Frontiers in Neuroscience 2018, 12, 632, 10.3389/fnins.2018.00632.
- David W. Killilea; Hani Atamna; Charles Liao; Bruce N. Ames; Iron Accumulation During Cellular Senescence in Human FibroblastsIn Vitro. Antioxidants & Redox Signaling 2003, 5, 507-516, 10.1089/152308603770310158.
- Shashank Masaldan; Sharnel A.S. Clatworthy; Cristina Gamell; Peter M. Meggyesy; Antonia-Tonia Rigopoulos; Sue Haupt; Ygal Haupt; Delphine Denoyer; Paul A. Adlard; Ashley Bush; et al.Michael A. Cater Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biology 2017, 14, 100-115, 10.1016/j.redox.2017.08.015.
- Maryam Mazhar; Ahmad Ud Din; Hamid Ali; Guoqiang Yang; Wei Ren; Li Wang; Xiaohui Fan; Sijin Yang; Implication of ferroptosis in aging. Cell Death Discovery 2021, 7, 1-9, 10.1038/s41420-021-00553-6.
- Alexandra Moreno-García; Alejandra Kun; Olga Calero; Miguel Medina; Miguel Calero; An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Frontiers in Neuroscience 2018, 12, 464, 10.3389/fnins.2018.00464.
- Annika Höhn; Tobias Jung; Stefanie Grimm; Tilman Grune; Lipofuscin-bound iron is a major intracellular source of oxidants: Role in senescent cells. Free Radical Biology and Medicine 2010, 48, 1100-1108, 10.1016/j.freeradbiomed.2010.01.030.
- Chendong Pan; Kalpita Banerjee; Guillermo L. Lehmann; Dena Almeida; Katherine A. Hajjar; Ignacio Benedicto; Zhichun Jiang; Roxana A. Radu; David H. Thompson; Enrique Rodriguez-Boulan; et al.Marcelo M. Nociari Lipofuscin causes atypical necroptosis through lysosomal membrane permeabilization. Proceedings of the National Academy of Sciences 2021, 118, 47, 10.1073/pnas.2100122118.
- Angélique Chevriaux; Thomas Pilot; Valentin Derangère; Harmonie Simonin; Pierre Martine; Fanny Chalmin; François Ghiringhelli; Cédric Rébé; Cathepsin B Is Required for NLRP3 Inflammasome Activation in Macrophages, Through NLRP3 Interaction. Frontiers in Cell and Developmental Biology 2020, 8, 167, 10.3389/fcell.2020.00167.
- Raquel Gómez-Sintes; María Dolores Ledesma; Patricia Boya; Lysosomal cell death mechanisms in aging. Ageing Research Reviews 2016, 32, 150-168, 10.1016/j.arr.2016.02.009.
- Sheng-Yu Zhu; Ren-Qi Yao; Yu-Xuan Li; Peng-Yue Zhao; Chao Ren; Xiao-Hui Du; Yong-Ming Yao; Lysosomal quality control of cell fate: a novel therapeutic target for human diseases. Cell Death & Disease 2020, 11, 1-13, 10.1038/s41419-020-03032-5.
- Xiandi Zhu; Yn Sun; Di Chen; Jingfeng Li; Xia Dong; Jie Wang; Huaiwen Chen; Ying Wang; Fulei Zhang; Jinaxin Dai; et al.Rogério P. PirracoShangjing GuoAlexandra P. MarquesRui L. ReisWei Li Mastocarcinoma therapy synergistically promoted by lysosome dependent apoptosis specifically evoked by 5-Fu@nanogel system with passive targeting and pH activatable dual function. Journal of Controlled Release 2017, 254, 107-118, 10.1016/j.jconrel.2017.03.038.
- Chunpeng Gao; Yue Ding; Laifu Zhong; Liping Jiang; Chengyan Geng; Xiaofeng Yao; Jun Cao; Tacrine induces apoptosis through lysosome- and mitochondria-dependent pathway in HepG2 cells. Toxicology in Vitro 2014, 28, 667-674, 10.1016/j.tiv.2014.02.001.
- Shuo Liu; Yun Li; Harry M. C. Choi; Chinmoy Sarkar; Eugene Y. Koh; Junfang Wu; Marta M. Lipinski; Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis. Cell Death & Disease 2018, 9, 1-14, 10.1038/s41419-018-0469-1.
- Jian Zou; Taro Kawai; Tetsuo Tsuchida; Tatsuya Kozaki; Hiroki Tanaka; Kyung-Sue Shin; Himanshu Kumar; Shizuo Akira; Poly IC Triggers a Cathepsin D- and IPS-1-Dependent Pathway to Enhance Cytokine Production and Mediate Dendritic Cell Necroptosis. Immunity 2013, 38, 717-728, 10.1016/j.immuni.2012.12.007.
- Huan Gao; Yuansong Bai; Yuanyuan Jia; Yanan Zhao; Rui Kang; Daolin Tang; Enyong Dai; Ferroptosis is a lysosomal cell death process. Biochemical and Biophysical Research Communications 2018, 503, 1550-1556, 10.1016/j.bbrc.2018.07.078.
- Timothy Nacarelli; Ashley Azar; Christian Sell; Aberrant mTOR activation in senescence and aging: A mitochondrial stress response?. Experimental Gerontology 2014, 68, 66-70, 10.1016/j.exger.2014.11.004.
- Giovanni Stallone; Barbara Infante; Concetta Prisciandaro; Giuseppe Grandaliano; mTOR and Aging: An Old Fashioned Dress. International Journal of Molecular Sciences 2019, 20, 2774, 10.3390/ijms20112774.
- Hong Zhang; Henry Hoff; Theresa Marinucci; Vincent J. Cristofalo; Christian Sell; Mitogen-Independent Phosphorylation of S6K1 and Decreased Ribosomal S6 Phosphorylation in Senescent Human Fibroblasts. Experimental Cell Research 2000, 259, 284-292, 10.1006/excr.2000.4965.
- Manuela Barilari; Grégory Bonfils; Caroline Treins; Vonda Koka; Delphine De Villeneuve; Sylvie Fabrega; Mario Pende; ZRF 1 is a novel S6 kinase substrate that drives the senescence programme. The EMBO Journal 2017, 36, 736-750, 10.15252/embj.201694966.
- M. Sandri; L. Barberi; A. Y. Bijlsma; B. Blaauw; K. A. Dyar; G. Milan; C. Mammucari; C. G. M. Meskers; G. Pallafacchina; A. Paoli; et al.D. PionM. RoceriV. RomanelloA. L. SerranoL. TonioloL. LarssonA. B. MaierP. Muñoz-CánovesA. MusaròM. PendeC. ReggianiR. RizzutoS. Schiaffino Signalling pathways regulating muscle mass in ageing skeletal muscle. The role of the IGF1-Akt-mTOR-FoxO pathway. Biogerontology 2013, 14, 303-323, 10.1007/s10522-013-9432-9.
- Yong-Xin Sun; Xunming Ji; XiaoOu Mao; Lin Xie; Jianping Jia; Veronica Galvan; David A. Greenberg; Kunlin Jin; Differential Activation of mTOR Complex 1 Signaling in Human Brain with Mild to Severe Alzheimer's Disease. Journal of Alzheimer's Disease 2013, 38, 437-444, 10.3233/JAD-131124.
- Remi-Martin Laberge; Yu Sun; Arturo V. Orjalo; Christopher K. Patil; Adam Freund; Lili Zhou; Samuel C. Curran; Albert R. Davalos; Kathleen A. Wilson-Edell; Su Liu; et al.Chandani LimbadMarco DemariaPatrick LiGene B. HubbardYuji IkenoMartin A JavorsPierre-Yves DesprezChristopher C. BenzPankaj KapahiPeter S. NelsonJudith Campisi MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nature Cell Biology 2015, 17, 1049-1061, 10.1038/ncb3195.
- Masako Narita; Andrew R. J. Young; Satoko Arakawa; Shamith A. Samarajiwa; Takayuki Nakashima; Sei Yoshida; Sungki Hong; Lorraine S. Berry; Stefanie Reichelt; Manuela Ferreira; et al.Simon TavaréKen InokiShigeomi ShimizuMasashi Narita Spatial Coupling of mTOR and Autophagy Augments Secretory Phenotypes. Science 2011, 332, 966-970, 10.1126/science.1205407.
- Eng-Soon Khor; Pooi-Fong Wong; The roles of MTOR and miRNAs in endothelial cell senescence. Biogerontology 2020, 21, 517-530, 10.1007/s10522-020-09876-w.
- Chung-Han Lee; Ken Inoki; Magdalena Karbowniczek; Emmanuel Petroulakis; Nahum Sonenberg; Elizabeth Petri Henske; Kun-Liang Guan; Constitutive mTOR activation in TSC mutants sensitizes cells to energy starvation and genomic damage via p53. The EMBO Journal 2007, 26, 4812-4823, 10.1038/sj.emboj.7601900.
- Eng-Soon Khor; Pooi-Fong Wong; Endothelial replicative senescence delayed by the inhibition of MTORC1 signaling involves MicroRNA-107. The International Journal of Biochemistry & Cell Biology 2018, 101, 64-73, 10.1016/j.biocel.2018.05.016.
- Andrea Alimonti; Caterina Nardella; Zhenbang Chen; John Clohessy; Arkaitz Carracedo; Lloyd C. Trotman; Ke Cheng; Shohreh Varmeh; Sara C. Kozma; George Thomas; et al.Erika RosivatzRudiger WoscholskiFrancesco CognettiHoward I. ScherPier Paolo Pandolfi A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. Journal of Clinical Investigation 2010, 120, 681-693, 10.1172/jci40535.
- Seung Hee Jung; Hyun Jung Hwang; Donghee Kang; Hyun A. Park; Hyung Chul Lee; Daecheol Jeong; Keunwook Lee; Heon Joo Park; Young-Gyu Ko; Jae-Seon Lee; et al. mTOR kinase leads to PTEN-loss-induced cellular senescence by phosphorylating p53. Oncogene 2018, 38, 1639-1650, 10.1038/s41388-018-0521-8.
- Olena Kucheryavenko; Glyn Nelson; Thomas von Zglinicki; Viktor I. Korolchuk; Bernadette Carroll; The mTORC1-autophagy pathway is a target for senescent cell elimination. Biogerontology 2019, 20, 331-335, 10.1007/s10522-019-09802-9.
- Leena P. Bharath; Madhur Agrawal; Grace McCambridge; Dequina A. Nicholas; Hatice Hasturk; Jing Liu; Kai Jiang; Rui Liu; Zhenheng Guo; Jude Deeney; et al.Caroline M. ApovianJennifer Snyder-CappioneGregory S. HawkRebecca M. FleemanRiley M.F. PihlKatherine ThompsonAnna C. BelkinaLicong CuiElizabeth A. ProctorPhilip A. KernBarbara S. Nikolajczyk Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metabolism 2020, 32, 44-55.e6, 10.1016/j.cmet.2020.04.015.
- Zoya N. Demidenko; Svetlana G. Zubova; Elena I. Bukreeva; Valery A. Pospelov; Tatiana V. Pospelova; Mikhail V. Blagosklonny; Rapamycin decelerates cellular senescence. Cell Cycle 2009, 8, 1888-1895, 10.4161/cc.8.12.8606.
- Olga V. Leontieva; Zoya N. Demidenko; Mikhail V. Blagosklonny; Contact inhibition and high cell density deactivate the mammalian target of rapamycin pathway, thus suppressing the senescence program. Proceedings of the National Academy of Sciences 2014, 111, 8832-8837, 10.1073/pnas.1405723111.
- Ju-Xian Song; Yue-Ru Sun; Ivana Peluso; Yu Zeng; Xing Yu; Jia-Hong Lu; Zheng Xu; Ming-Zhong Wang; Liang-Feng Liu; Ying-Yu Huang; et al.Lei-Lei ChenSiva Sundara Kumar DurairajanHong-Jie ZhangBo ZhouAiping LuAndrea BallabioDiego L. MedinaZhihong GuoMin Li A novel curcumin analog binds to and activates TFEB in vitro and in vivo independent of MTOR inhibition. Authophagy 2016, 12, 8, 10.6084/m9.figshare.3376261.
- Taiji Tsunemi; Travis D. Ashe; Bradley E. Morrison; Kathryn R. Soriano; Jonathan Au; Ruben A. Vázquez Roque; Eduardo R. Lazarowski; Vincent A. Damian; Eliezer Masliah; Albert R. La Spada; et al. PGC-1α Rescues Huntington’s Disease Proteotoxicity by Preventing Oxidative Stress and Promoting TFEB Function. Science Translational Medicine 2012, 4, 142ra97-142ra97, 10.1126/scitranslmed.3003799.
- Mickael Decressac; Bengt Mattsson; Pia Weikop; Martin Lundblad; Johan Jakobsson; Anders Björklund; TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proceedings of the National Academy of Sciences 2013, 110, E1817-26, 10.1073/pnas.1305623110.
- Benjamin Dehay; Jordi Bové; Natalia Rodriguez-Muela; Celine Perier; Ariadna Recasens; Patricia Boya; Miquel Vila; Pathogenic Lysosomal Depletion in Parkinson's Disease. The Journal of Neuroscience 2010, 30, 12535-12544, 10.1523/jneurosci.1920-10.2010.
- Kiri Kilpatrick; Yimeng Zeng; Tommy Hancock; Laura Segatori; Genetic and Chemical Activation of TFEB Mediates Clearance of Aggregated α-Synuclein. PLOS ONE 2015, 10, e0120819-e0120819, 10.1371/journal.pone.0120819.
- Yusheng Cai; Huanhuan Zhou; Yinhua Zhu; Qi Sun; Yin Ji; Anqi Xue; Yuting Wang; Wenhan Chen; Xiaojie Yu; Longteng Wang; et al.Han ChenCheng LiTuoping LuoHongkui Deng Elimination of senescent cells by β-galactosidase-targeted prodrug attenuates inflammation and restores physical function in aged mice. Cell Research 2020, 30, 574-589, 10.1038/s41422-020-0314-9.