Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Josef Gillson.
Pancreatic cancer is known to have the lowest survival outcomes among all major cancers, and unfortunately, this has only been marginally improved over last four decades. The innate characteristics of pancreatic cancer include an aggressive and fast-growing nature from powerful driver mutations, a highly defensive tumor microenvironment and the upregulation of advantageous survival pathways such as autophagy. Autophagy involves targeted degradation of proteins and organelles to provide a secondary source of cellular supplies to maintain cell growth.
- pancreatic ductal adenocarcinoma
- autophagy
- tumor microenvironment
1. Autophagy
1.1. Autophagy Types and Selectivity
The degradation of cellular contents is a central process in all eukaryotic cells [22[1][2],23], which is primarily performed by the ubiquitin-proteasomal system (UPS) and autophagy [17,23,24][2][3][4]. The UPS is a highly specialized mechanism that targets old, dysfunctional or unwanted cellular material through ubiquitination and degrades the content into smaller molecular units [17,19,24][3][4][5]. Autophagy features a more versatile targeting spectrum as it can incorporate organelles and a more diverse range of proteins than UPS [18,24,25][4][6][7].
Autophagic activity can be executed by three main mechanisms: chaperone-mediated autophagy (CMA), microautophagy and macroautophagy [26,27,28][8][9][10]. CMA is a highly specific process and relies on the recognition of unique targeting motifs located on certain cytosolic substrates by a cytoplasmic chaperone, such as Hsc70, which leads to their delivery to lysosomes for degradation [29,30][11][12]. Microautophagy possesses both specific and non-specific targeting and involves the direct invagination of targets into lysosomes [31][13]. The mechanism underlying macroautophagy is characterized by the de novo formation of phagophores around cytoplasmic structural mass, which matures into an autophagosome that fuses with a lysosome to allow the localized hydrolases to degrade the target protein or organelle into smaller, useable molecules [18,32,33,34][6][14][15][16] (Figure 1).
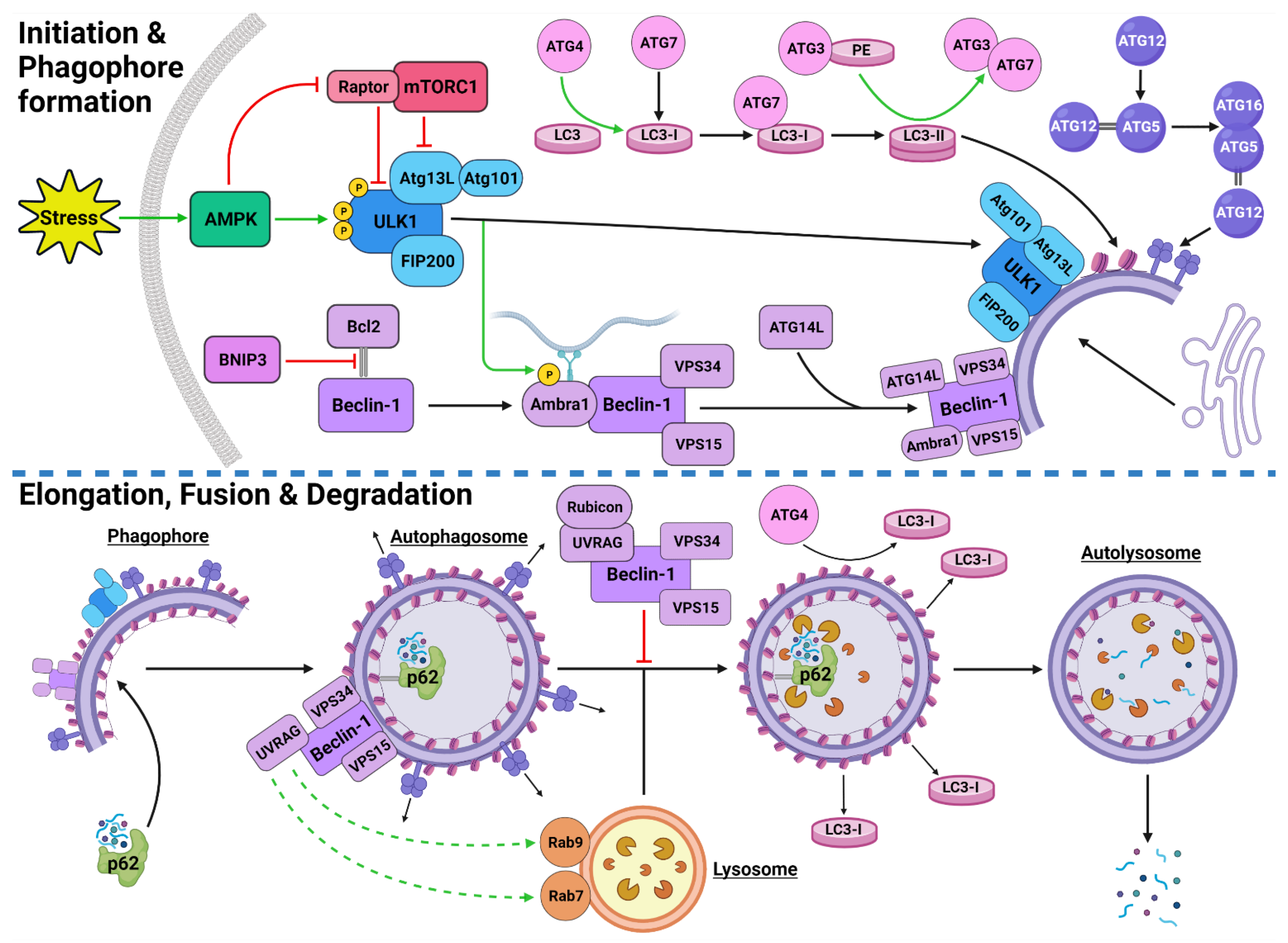
Figure 1. Stress-Induced Autophagy Pathway and Machinery. Tumor microenvironmental stress stimulates autophagy via AMPK activation which induces autophagic initiation. The ULK1 complex and PI3KC3-C1 facilitate phagophore formation which matures and elongates into an autophagosome by structural proteins LC3-II and the ATG5-ATG12-ATG16 complex. The autophagosome forms around the target protein/organelle and fuses with a lysosome mediated by the PI3KC3-C2. The cargo is degraded into various biomolecules and released into the cytoplasm. Black arrows indicate binding to or moving to, green arrows indicate activation, green dashed arrows indicate attraction, red arrows indicate inhibition. Created with BioRender.com (accessed on 24 February 2022).
Autophagy exhibits both selective and non-selective targeting of cytoplasmic contents [35,36,37,38][17][18][19][20]. While these mechanisms use the same intracellular core machinery, selective targeting utilizes a range of specialized receptors and chaperones [39][21]. The autophagic chaperone p62 is known to sequester the targeted protein/organelle and carries it to a receptor on the autophagosome for degradation [38,39][20][21]. Additionally, non-selective autophagy of small cytoplasmic proteins is more prominent under normal conditions and during early stages of stressful starvation events [40][22]. In contrast, prolonged stressful starvation triggers a rise in specific autophagic targeting of more complex proteins and organelles [40][22]. This indicates that stress can instigate a stronger, more selective response. Some examples of autophagic selectivity include pexophagy (peroxisomes) [41][23], mitophagy (mitochondria) [42,43][24][25] and xenophagy (bacteria during an infection) [44,45][26][27].
The process of autophagy occurs in all cell types and is an integral part of homeostatic regulation throughout the cellular lifecycle [46][28]. However, autophagy is well established as a stress-responsive process that is highly upregulated during starved conditions to generate more energy and nutrients [47[29][30][31],48,49], cellular remodeling from growth and development [50[32][33],51], and increased during oxidative stress [37,52][19][34].
1.2. Autophagy Process and Machinery
1.2.1. Autophagy Initiation
The initiation of autophagic flux is regulated by two important protein complexes, namely, UNC-51-like kinase (ULK1) complex and phosphoinositide 3-kinase class III-complex 1 (PI3KC3-C1) (Figure 1). When phosphorylated by its upstream regulators, ULK1 can bind to both the focal adhesion kinase family interacting protein of 200 kDa (FIP200) and the conjugate of autophagy-related protein 13 (ATG13) and ATG101 to form the ULK1-FIP200-ATG13-ATG101 complex, which is also known as the ULK1 initiation complex [53,54][35][36]. The ULK1 initiation complex is vital for the completion of autophagy and its inhibition was shown to significantly reduce autophagic initiation and prevent cell survival under nutrient-deprived conditions [55,56][37][38]. Upon its formation, the ULK1 initiation complex triggers an array of downstream signaling pathways to begin the formation of an isolation membrane, known as a phagophore [57][39]. The most significant of these signals involves the activation of PI3KC3-C1 [57,58][39][40]. This ULK1-mediated phosphorylation of Beclin-1 can be enhanced by both ATG14-like (ATG14L) and ultraviolet radiation resistance-associated gene protein (UVRAG) [57][39].
Inactive Beclin-1 is bound to B-cell lymphoma 2 (Bcl-2) [59][41] (Figure 2). When released by other competitive BH-3-binding proteins, such as BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) (Figure 2), Beclin-1 binds to vacuolar protein sorting 34 (VPS34), VPS15 and, autophagy and Beclin-1 regulator 1 (AMBRA1), which anchors the complex to microtubular dynein [58,60][40][42] (Figure 1). ULK1 activates the PI3KC3-C1 by: (1) phosphorylating AMBRA1 to release it from the dynein; and (2) phosphorylating both Beclin-1 and VPS34 allowing ATG14L to bind [58,61,62,63][40][43][44][45]. The activated ULK1 complex and PI3KC3-C1 then localize to the isolation membrane on the ER/golgi apparatus [64,65][46][47].
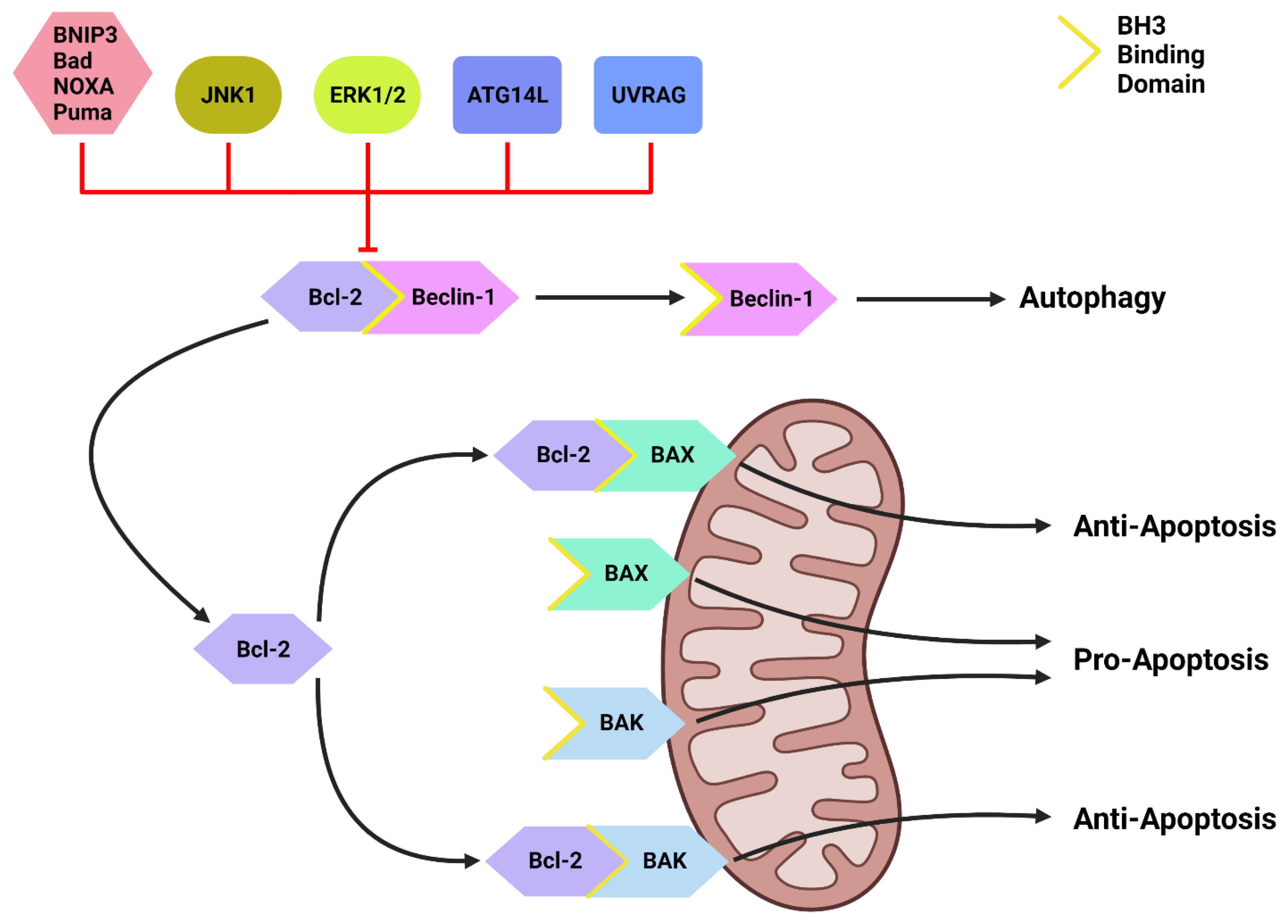
Figure 2. The Bcl-2/Beclin-1 Interaction. Bcl-2 family proteins (BNIP3, Bad, NOXA, Puma) that compete for the BH3 binding site and other proteins such as JNK1, ATG14L and UVRAG can disrupt the Bcl-2/Beclin-1 complex. This disruption frees Beclin-1 to form the PI3KC-C1 and initiate autophagic initiation or PI3KC-C2 to promote autolysosome fusion. Free Bcl-2 can also bind to the BH3-binding site on BAX and BAK to protect the mitochondria and suppress apoptotic function. Created with BioRender.com (accessed on 4 March 2022).
1.2.2. Autophagosome Formation
The PI3KC3-C1 and ULK1 complex facilitate phagophore elongation into an autophagosome, which is characterized by two ubiquitin-like systems, namely, ATG5-ATG12-ATG16 conjugate and microtubule-associated proteins 1A/1B light chain 3A (LC3) [60,66][42][48] (Figure 1). ATG5 forms a covalent bond with ATG12 which is later joined by ATG16 [67,68][49][50]. LC3 is converted to LC3-I by ATG4, which stimulates the binding of ATG7 to attract ATG3 resulting in the ligation of phosphatidylethanolamine (PE) with LC3-I to form LC3-PE conjugate (i.e., LC3-II) [32,69,70][14][51][52]. A multitude of LC3-II and ATG5-ATG12-ATG16 complexes then localise to the phagophore to begin the formation of the autophagosome [68][50] (Figure 1).
1.2.3. Autophagosome Maturation
The maturation process depends upon interactions between LC3-II and sequesterome-1 (p62), which are also two distinct markers of autophagic flux [71][53] (Figure 1). LC3 is one of three human homologs of Atg8 in yeast, the other two are GABARAP and GATE-16, both of which function similar to LC3 [72][54]. Similarly, p62 shares its role with the homologs BNIP3L, NBR1 and Alfy [73,74,75][55][56][57]. p62 provides selectivity to the autophagic process by recognizing ubiquitinated target proteins and sequestering the target towards the phagophore [18,73][6][55]. It then binds directly to the internal membrane-bound LC3-II using the LC3 recognitions sequence in a ligand-receptor-like manner, which then stabilizes the target protein in place to allow the autophagosome to form around it [39,76][21][58]. Upon autophagosome formation, external LC3-II remains on the surface of the membrane, the internal LC3-II and p62 are enclosed within the membrane, while the ATG5-ATG12-ATG16 complex progressively detaches [77][59] (Figure 1). Interestingly, ATG3 knockouts generated autophagosomes lacking LC3-II that were still able to bind to p62 and complete autophagic flux, suggesting that the ATG5-ATG12-ATG16 complex was able to attract p62 independently of LC3-II [39][21]. This result supports the concept that autophagy is a tightly regulated process with complex machinery that can adapt and respond to various forms of disruption throughout the process.
1.2.4. Autolysosome Formation and Cargo Degradation
Once autophagosomes have fully enveloped their target, they fuse with lysosomes to form autolysosomes [78][60]. Interestingly, UVRAG competes with ATG14L for the same binding site on Beclin-1 [62][44]. With UVRAG bound, this complex is known as the PI3KC3-C2 and facilitates the attraction of the lysosome-bound GTPases, Rab7 and Rab9 to the autophagosome [62,79,80,81][44][61][62][63] (Figure 1). Rubicon can bind to UVRAG to mediate a suppressive effect on autolysosome formation through the interference with Rab7 attraction [82,83][64][65]. However, it was demonstrated that in circumstances of sustained autophagic activation, the UVRAG expression levels outnumber the Rubicon expression, and therefore, manages to maintain dominance of Rab7 activation and trigger autolysosome formation [83][65].
Upon autolysosome formation, the acidic hydrolases and proteases from the lysosome target the contents and remaining membranous proteins which causes proteolysis to yield smaller products such as amino acids, peptides and free fatty acids [84][66] (Figure 1). These products are released into the cytoplasm to be reused, excreted into the bloodstream for use elsewhere, restore the intracellular free amino acid pool or directly transported to the ribosome for protein synthesis [84,85][66][67]. The degradation of internally bound LC3-II and p62 is indicative of autophagic flux [86][68]. The externally bound LC3-II is not degraded, but delipidated by ATG4 into LC3-I, which can then be reused in the next autophagic cycle [72][54].
1.3. Upstream Autophagy Regulation
Autophagy regulation upstream of the core autophagic machinery involves numerous proteins and pathways that indirectly activate or inhibit this critical catabolic process. The major upstream pathways involved in regulation of autophagic machinery are: (i) PI3K class I (PI3KC1)/protein kinase B (AKT)/mammalian target of rapamycin complex 1 (mTORC1) pathway; (ii) mitogen activated protein kinase (MAPK) pathway; (iii) adenosine monophosphate-activated protein kinase (AMPK); and (iv) Bcl-2.
1.3.1. PI3K/AKT/mTORC1 Pathway
The activation of the PI3KC1 complex from either receptor tyrosine kinases or KRAS leads to constitutive phosphorylation of the phospholipid PIP2 into PIP3 [87][69] (Figure 3). Phosphatase and tensin homolog (PTEN) functions to reverse the action of PI3K by directly dephosphorylating PIP3 back into inactive PIP2 to maintain regulation of the pathway [88,89][70][71]. PIP3 activates AKT, which then phosphorylates tuberous sclerosis complex 2 (TSC2) at five different sites (Ser939, Ser981, Ser1130, Ser1132 and Thr1462) causing it to destabilize and dissociate from TSC1 [90,91][72][73]. This dissociation prevents the dual protein complex of TSC1 and TSC2 from inhibiting ras homolog enriched in brain (RHEB), a constitutive activator of mTORC1 [90][72]. mTORC1 is comprised of mTOR, GβL, PRAS40 and Raptor [92][74]. Sustained mTORC1 activity mediates autophagy suppression via the phosphorylation of the major initiation proteins ULK1 (Ser757) and ATG13L, rendering them inactive [53,54,93,94,95][35][36][75][76][77]. Therefore, the activation of PI3K/AKT/mTORC1 inhibits autophagy while the suppression of the PI3K/AKT/mTORC1 pathway promotes autophagy [87,96][69][78] (Figure 3). Additionally, mTORC1 operates to activate S6K1 and destabilizes the eIF-4E and 4E-BP1 complex to collectively promote protein synthesis [87,97][69][79], further reinforcing its significance in managing cellular protein synthesis or degradation.
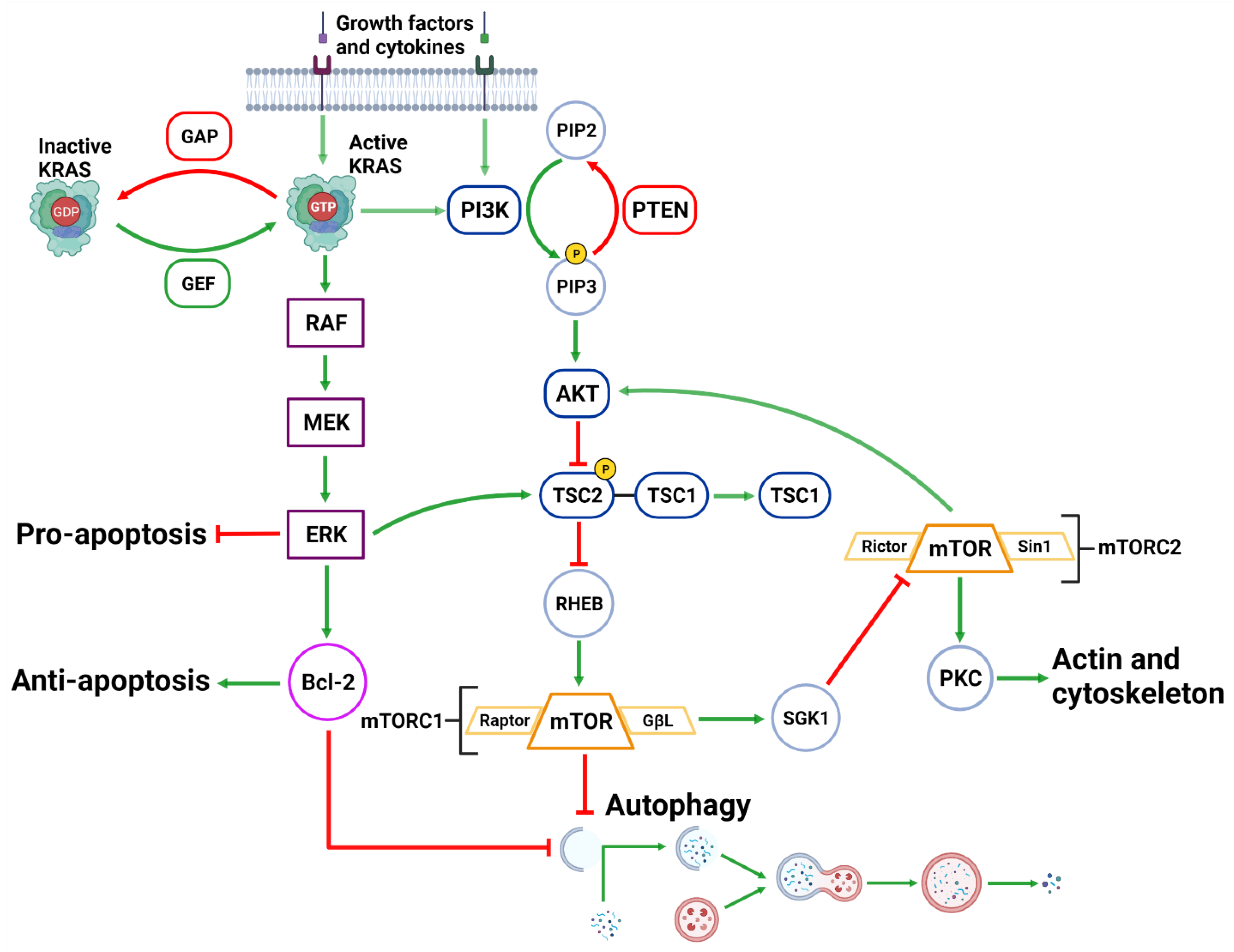
Figure 3. Upstream Autophagy Regulation. Extracellular growth factors and cytokines activate KRAS and PI3K. GAP and GEF regulate KRAS activity which begins the MAPK cascade of activating RAF, MEK and ERK. ERK can inhibit pro-apoptotic function and support anti-apoptotic function via Bcl-2. ERK can also inhibit mTORC1 which facilitates autophagic initiation. PI3K phosphorylates PIP3 which is regulated by PTEN dephosphorylation. PIP3 activates AKT causing the destabilization of TSC2-TSC1 complex. This supports mTORC1 activity and suppresses autophagic initiation. mTORC1 can also regulate AKT via a feedback loop and suppress cytoskeleton activity involving SGK1 and mTORC2. Created with BioRender.com (accessed on 28 March 2022).
1.3.2. MAPK Pathway
Downstream of KRAS, the rapid accelerated fibrosarcoma (RAF)/mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway operates parallel to the PI3K/AKT pathway and is similarly integral to growth signaling with a major influence on tumor onset and survival [98,99][80][81] (Figure 3). In addition to transcription-related nuclear effects and the regulation of cytosolic proteins [100][82], MEK and ERK1/2 are also known for their crosstalk into other pathways which allows for an increased range of effects on autophagy when compared to the PI3K/AKT pathway. This can be epitomized by ERK1/2 activation of TSC2 (at the ERK D domain) which leads to RHEB inhibition and subsequent mTORC1 destabilization [101][83] (Figure 3). This results in increased levels of Beclin-1 and ULK1 leading to significantly increased autophagic initiation [101,102][83][84]. Additionally, phosphorylation of Bcl-2 by ERK1/2 is shown to promote its dissociation from Beclin-1, resulting in autophagic induction [103][85] (Figure 2).
Interestingly, the strength of the MEK/ERK signal dictates the effectiveness of autophagy activity, such that moderate MEK/ERK activity-induced cyto-protective autophagy and sustained MEK/ERK activation can cause cyto-destructive autophagy [101][83]. ERK inhibition or ERK knockdown was unable to fully repress autophagic flux [101][83]. However, MEK inhibition was found to completely abrogate autophagic activity [101][83]. As ERK is one of the downstream MEK effector proteins, this result indicates that MEK was capable of bypassing ERK and used alternative effector proteins to sustain the stimulatory autophagic signal.
The effects of MAPK activity may be described as a more versatile and passive signaling pathway than a binary pathway. In liver and breast cancer, MEK inhibition (PD98059) completely suppressed rapamycin and serum starvation-induced autophagy observed from sustained mTORC1 activity and reduced Beclin-1 levels [101][83]. It has therefore been described that MEK/ERK activation is required for autophagy activation [104][86]. Interestingly, there is also evidence of a feedback network where autophagy related genes are capable of regulating ERK1/2 phosphorylation. Either MAP1LC3 or ATG5 mRNA silencing in mice resulted in reduced ERK phosphorylation and suppressed MAPK signaling [105][87].
1.3.3. AMPK
AMPK is a well-established upstream regulator of autophagy [54,71,106][36][53][88]. As a stress-responsive protein, AMPK reacts to decreased cellular energy and resource levels to stimulate survival pathways such as autophagy and glycolysis [106,107][88][89] (Figure 4). AMPK consists of a regulatory γ subunit, a structural β subunit and a catalytic α subunit [108][90]. Stress-associated AMPK activation occurs from a direct mechanism involving depleted adenosine triphosphate (ATP) levels that raise cytoplasmic adenosine mono/diphosphate (AMP/ADP) levels [109,110][91][92] (Figure 4). AMPK is also activated by three upstream regulators in response to different stimuli: (1) liver kinase B1 (LKB1), which responds to cellular energy levels; (2) Ca2+/calmodulin-dependent kinase kinase β (CaMKKß) activation by increased cytoplasmic calcium (Ca2+) from ER stress; and (3) transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1) [106,107,111,112][88][89][93][94] [113,114][95][96] (Figure 4).
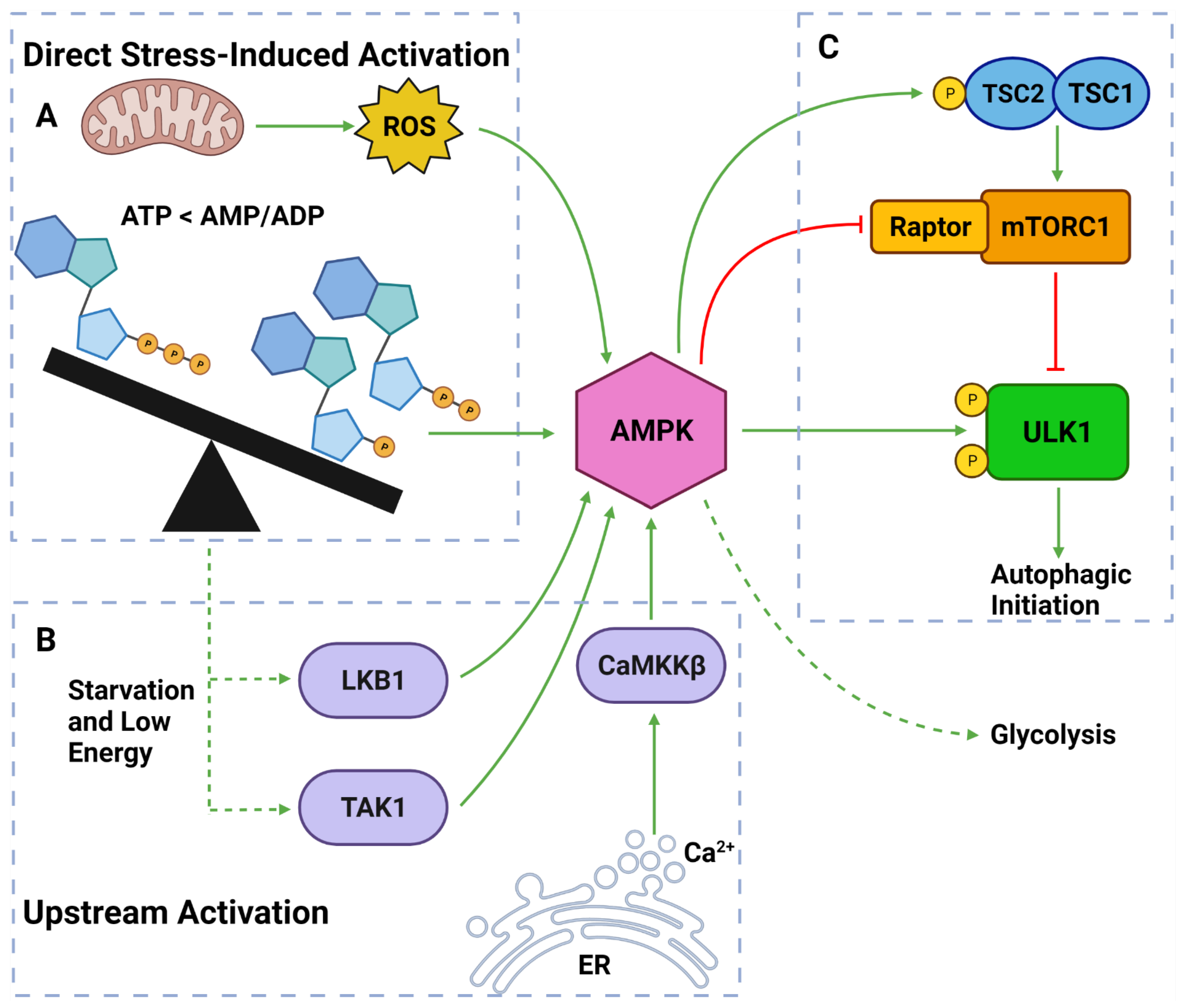
Figure 4. AMPK Regulators and Effectors. (A) AMPK can be directly activated by stress from: overworked/stressed mitochondria produced ROS; AMP and ADP due to decreased ATP levels. (B) AMPK can also be activated by upstream regulators: LKB1 and TAK1 which respond to reduced energy levels; CaMKKβ which responds to increased cytoplasmic calcium from ER stress. (C) Once activated, AMPK can induce autophagy by: phosphorylating TSC2; inhibiting Raptor on mTORC1; phosphorylating ULK1 at Ser317 and Ser777. Activated AMPK can also upregulate glycolytic activity. Created with BioRender.com (accessed on 6 April 2022).
Once activated, AMPK upregulates autophagy via from 3 major pathways: (1) phosphorylation and deactivation of Raptor (a protein within the mTORC1); (2) activation of TSC2, causing RHEB inhibition and subsequent mTORC1 inhibition; and (3) ULK1 phosphorylation at Ser317 and Ser777 sites [53,54,94,115][35][36][76][97] (Figure 4). It should also be noted that the activation of TSC2 can directly oppose the PI3K pathway-induced autophagic suppression [106,107,111][88][89][93]. Collectively, AMPK is a vital autophagic activator which has a complex, yet well understood, mechanism of activating autophagy.
1.3.4. Beclin-1 & Bcl-2
Another important regulator of autophagic initiation involves the Bcl-2 family of apoptosis-related proteins. The Bcl-2 protein itself typically exerts anti-apoptotic signaling where it binds to BH3 domains on pro-apoptotic proteins such as Bcl-2-associated X protein (BAX) and Bcl-2 homologous antagonist killer (BAK) to protect the mitochondria [116][98] (Figure 2). Beclin-1 also contains a BH3 binding domain and has been found bound to Bcl-2 in the form of a dual protein complex [59,117,118][41][99][100]. Importantly, the Beclin-1/Bcl-2 complex restrains Beclin-1 from initiating autophagy and prevents Bcl-2 from binding to pro-apoptotic proteins, leading to increased apoptosis [59,117,118][41][99][100] (Figure 2).
Beclin-1 can be dissociated from Bcl-2 via (1) JNK1; (2) other BH3 domain containing Bcl-2 family members, such as BNIP3, Bad, Noxa, Puma, etc.; and (3) other autophagy promoting proteins such as UVRAG and ATG14L [119,120][101][102] (Figure 2). This suggests that Bcl-2 plays a major role in the crosstalk between apoptotic and autophagic machinery.
2. Autophagy in Pancreatic Cancer Progression
There has been emerging evidence of a critical role played by the autophagic pathway in pancreatic cancer progression [213,222][103][104]. Notably, previous studies have shown that advanced/high grade PDAC have elevated autophagy when compared to normal pancreas or low grade PDAC [210,288][105][106]. This is understandable as protein synthesis is vital for the overstimulated growth and unlocking the metastatic potential of cancers [170][107]. Overall, increased autophagic upregulation in PDAC could be instigated by a combination of driver mutations and a highly stressful TME [15,147][108][109]. Autophagy may support stressed neoplastic cells directly by providing more biomaterials or by influencing alternate pathway to support tumor survival.2.1. Autophagic Regulation in PDAC
Autophagy is tightly regulated by a variety of upstream pathways that are often mutated in PDAC. The PI3K/AKT pathway mediates autophagic inhibition, while the MEK/ERK pathway is deemed essential for autophagy activation [87,101,289][69][83][110] (Figure 3). KRAS is at the helm of these varying pathways and is frequently mutated in PDAC [290][111]. While KRAS influences autophagic regulation, it has opposing downstream effectors. Therefore, the net increase to autophagy in PDAC is determined by a balance between upstream regulatory pathways, TME stress and other stress-related proteins such as AMPK and HIF-1/2, which adopt a more primary role at advanced stages [291][112]. The relationship between AMPK, autophagy and PDAC progression is an area of active research. AMPK is often activated from low cellular ATP levels and is regulated by upstream proteins, such as LKB1 and CaMKKß, and can directly promote autophagic initiation through different mechanisms [108,110,292][90][92][113] (Figure 4). Through these interactions, AMPK-induced autophagy is highly prevalent in stressed PDAC and is considered a fundamental component of PDAC survivability [93,106,190,191][75][88][114][115] (Figure 1). Similarly to AMPK, HIF-1/2 are activated by hypoxic conditions and can promote autophagy via the transcription of BNIP-3 [292][113]. Increased HIF activity further strengthens the survival abilities of stressed PDAC and encourages EMT and neoplastic migration [234][116]. The excessive ROS levels in stressed PDAC can be simultaneously damaging and supporting [189,195,201][117][118][119]. ROS can directly stimulate AMPK, mTOR and HIF-1α-mediated autophagy, indicating that PDAC tumors are still able to utilize damaging ROS to aid survival and progress to more advanced stages [292,293][113][120].2.2. Autophagy Promotes Pancreatic Tumor Progression
It has been established that autophagy acts as a tumor suppressor in early stages of PDAC development through the degradation of oncogenic proteins and resistance to apoptosis [294][121]. However, as the tumor becomes more advanced, autophagy is recorded at abnormally high levels where it operates as a survival pathway and promotes cell growth [294][121]. TME-induced stress typically inflicts biological responses that prevents cellular growth [173,175,176,177][122][123][124][125]. However, autophagic upregulation can help aggressive PDAC adapt to the harsh conditions [210][105]. One of these mechanisms involves autophagic activity opposing apoptotic activity. Beclin-1-mediated autophagic initiation is positively correlated with anti-apoptotic Bcl-2 function [118][100] (Figure 2). Therefore, as autophagy remains activated, apoptotic activity is reduced. This can potentially result in increased tumor survival under TME stress, which provides tumors more time to grow and metastasize; and could suppress cytotoxic chemotherapies from stimulating apoptosis-induced cell death. PDAC chemotherapy is largely ineffective due to the protective stroma and can be further suppressed if tumor cells are actively opposing apoptosis [118,295][100][126]. This interaction could also explain why combination therapy involving autophagy inhibition is highly synergistic [179,246,291][112][127][128]. Importantly, cytoplasmic contents deemed unnecessary for tumor proliferation can be degraded into amino and fatty acids and boost the available pool [85][67]. Free amino acids can be transported into the ER and ribosomes to produce more vital proteins involved in cellular metabolism and cell division [85,296][67][129]. To maintain an increased growth rate, PDAC cells can upregulate glycolysis via the Warburg effect [13,225][130][131]. This increased activity demands more proteins to execute and is therefore, fueled by autophagic degradation [225][131]. Moreover, in the TCA cycle glutamine is one of the primary sources of carbon [297][132]. Notably, autophagy has been described as a major source for intracellular glutamine and hence, can directly support oxidative phosphorylation [298][133]. Autophagy is essential to PSC and CAF function since it can provide alanine for neighboring tumor cells and enhance the deposition of ECM proteins such as glycoproteins, collagen and elastin; and MMPs which increase the ECM remodeling [261,273][134][135]. Increased biomaterial availability can enhance the production of a range of proteins involved in various cellular functions. This could include actin, myosin and other cytoskeletal proteins to increase cell motility and promote cellular breakaway [299][136]. Autophagy has been observed targeting and degrading MHC-I in PDAC resulting in reduced levels [287][137]. Due to the importance of MHC-I in immune surveillance, this degradation can protect the tumor cells and can lead to uncontrolled tumor growth [287][137]. A recent study has also shown importance of autophagic induction in Schwann cell could promote perineural invasion, which is one of well-known poor prognostic factor in PDAC progression [274][138]. Current studies examining the relationship between autophagy and pancreatic cancer progression have shown critical importance of this pathway in tumor progression and its potential to be developed as a key therapeutic target for this aggressive disease. Future studies will offer further insights on the complexity of autophagy regulation, its importance to PDAC survival, and how it may be manipulated to provide a therapeutic advantage over the disease.2.3. The Role of Autophagy in Pancreatic Cancer Metastasis
Cancer metastasis is the main cause of cancer-related death in PDAC and is therefore, a crucial area to be investigated [300,301,302,303][139][140][141][142]. PDAC is often characterized by its early metastatic features, resistance to anti-cancer therapies and poor prognosis [1,304][143][144]. Emerging evidence implies that the role of autophagy in cancer progression is complex, and often multifaceted, as contrasting studies suggest that it can be metastasis-promoting or suppressing depending on the stage of the disease, different tumor types and involves other pathway interactions [305,306][145][146].2.3.1. Autophagy as a Metastasis Promoter in Pancreatic Cancer
Most literature regarding PDAC establishes autophagy at a metastasis promoter. As a stress-induced pathway, it is known for maintaining cell survival and promoting the hallmarks of cancer, including metastasis [170,210][105][107]. Autophagy directly promotes metastasis through the degradation of proteins involved in focal adhesion. Paxillin is a binding protein that acts a scaffold for the recruitment of other proteins, such as focal adhesion kinase, and is responsible for binding actin in the cytoskeleton and extracellular integrin to create an anchor between cells and the ECM [307][147]. Autophagy was shown to degrade paxillin resulting in a reduced structural binding between tumor cells and the ECM, thus increasing neoplastic migration [308,309][148][149]. A further study using chloroquine (CQ) treatment in breast cancer models demonstrated a reduced rate of paxillin degradation both in vitro and in vivo [308][148]. More recently, this interaction has been confirmed in PDAC using a nano-bomb combination of gemcitabine and CQ. This combination was more effective at inhibiting paxillin degradation and downregulating MMP-2 when compared with either mono-treatment [246][128]. These results in both pancreatic and breast cancer models demonstrate that the autophagic degradation of paxillin led to increased metastatic potential. Hypoxia-induced autophagy is prominent in PDAC due to the advanced and stressed nature of the neoplasm (discussed in Section 4.2). Intermittent hypoxia was not only shown to upregulate autophagy-related proteins (Beclin-1 and LC3-II), but also increased EMT-related markers (vimentin and N-cadherin) and reduced the level of the cell-to-cell adhering protein, E-cadherin [233][150]. These latter findings were demonstrated to be due to the induction of hypoxia-induced autophagy [233][150]. In another set of studies, the metastasis suppressor, N-myc downstream regulator gene 1 (NDRG1) was shown to inhibit basal and hypoxia-induced autophagy via a dual-inhibitory mechanism involving impaired autophagic degradation and autolysosome formation in PDAC cells [49,310][31][151]. This inhibitory effect of NDRG1 on autophagy was shown to be mediated by suppression of PERK-eIF2α pathway [310][151]. Furthermore, NDRG1-mediated suppression via the PERK-eIF2α pathway was found to reduce migration [311][152]. Collectively, these studies demonstrate that upregulated autophagy in stressed PDAC is a metastasis promoter due to the targeted degradation of crucial proteins required to maintain cell to cell contact and upregulation of EMT marker levels. With the majority of PDAC patients exhibiting KRAS mutations [312][153], its relationship with the autophagic sequestering protein, p62, is also considered to support metastasis and is highly associated with poor prognosis [313,314][154][155]. The recorded high levels of p62 in PDAC can be attributed to the KRAS activation of NF-κB, which transcriptionally induces gene encoding SQSTM1 to produce p62 [315][156]. p62 was also found to maintain NF-κB activity through a feedforward loop [315][156]. As NF-κB transcriptional activity is vital for tumor invasion, EMT and anti-apoptosis [316[157][158],317], the study by Ling et al. implicates p62, and subsequently autophagy, as a major promoter of metastasis [315][156]. Another important feature of PDAC is the presence of cancer stem cells (CSCs). CSCs are characterized by their unique properties of self-renewal, sphere forming capacity and de-differentiation states, which contributes to and serves as a basis to cancer metastasis [318,319][159][160]. Rausch et al. showed that higher levels of CSC markers correlated with upregulated autophagy in PDAC [320][161]. Interestingly, autophagy inhibition in pancreatic CSCs resulted in apoptotic cell death and a reduction in migration and tumorigenicity [320][161]. Hypoxia is a crucial component of autophagic activation, metastasis and supports invasive stem cell-like features in PDAC cell lines [233,321][150][162]. Notably, CD133+ pancreatic CSCs were found to be colocalized to the hypoxic region within PDAC tumors [233][150]. Another study by Yang et al. further supported this hypothesis by positively correlating LC3 expression with the expression of CSC markers, aldehyde dehydrogenase 1 (ALDH1), CD44 and CD133 in PDAC tissues [322][163]. High co-expression of LC3/ALDH1 was associated with both poor overall survival and progression-free survival [322][163]. Indeed, the inhibition of autophagy by silencing ATG5, ATG7 and BECN1 or the administration of CQ significantly reduced pancreatic CSC population and activity [322][163]. These results suggest that stress-induced autophagy supports metastasis through the sustenance of pancreatic CSCs.2.3.2. Autophagy as a Metastasis Suppressor in Pancreatic Cancer
Where the previous studies demonstrate autophagy as a metastasis promoter, there are also studies that suggest an opposing effect. For instance, Akar et al. found that the elevated expression of the tissue transglutaminase, TG2, has been implicated in increased drug resistance, supporting metastatic phenotypes and poor patient prognosis in PDAC [323][164]. More specifically, TG2 increases EMT markers (vimentin, N-cadherin and fibronectin) and decreases E-cadherin levels [323][164]. The inhibition of protein kinase C-delta (PKCδ), which is vital for TG2 expression, resulted in excessive autophagic activation and Beclin-1-mediated cell death [323][164]. This result indicates that TG2-mediated autophagy suppression supports metastasis and implicates that autophagic activity suppresses metastasis. Studies demonstrating autophagic interactions that the partial (heterozygous deletion) or complete (homozygous deletion) loss of certain autophagy genes, have been shown to lead to contrasting outcomes. For instance, ATG5, a crucial protein in autophagosome formation, appears to contribute to metastatic capabilities in PDAC. Notably, there was a clear phenotypic difference between the complete and partial loss of ATG5 in autophagy-proficient transgenic mice with KRASG12D PDAC [324][165]. The homozygous knockout of ATG5 in mice harboring KRASG12D supported tumor initiation but prevented PDAC tumors from progressing into more malignant states [324][165]. Whereas, the heterozygous knockout of ATG5 in the same mouse model increased tumor incidence, malignancy and metastatic potential in PDAC by enhancing neoplastic migration and invasion when compared to the homozygous ATG knockout or KRASG12D control mice [324][165]. This relationship could be attributed to the numerous non-canonical autophagy-associated and intracellular degradation pathways that are responsible for the compensatory switch for the loss of ATG5, or as a protective mechanism exerted by PDAC cells. Therefore, this study demonstrates that partial loss of autophagy led a highly metastatic phenotype compared to mice with completely deficient or proficient autophagic activity. Collectively, the different models used in these studies suggest that autophagy plays both a pro- and anti-metastatic role in PDAC. This is presumably due to the diverse role of the molecules and proteins involved in autophagic regulation and thus, indicates that these interactions require careful consideration throughout the development of PDAC chemotherapeutic strategies that involve the autophagic pathway.References
- Nedelsky, N.B.; Todd, P.K.; Taylor, J.P. Autophagy and the ubiquitin-proteasome system: Collaborators in neuroprotection. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2008, 1782, 691–699.
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889.
- Korolchuk, V.I.; Menzies, F.M.; Rubinsztein, D.C. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2009, 584, 1393–1398.
- Ding, W.-X.; Ni, H.-M.; Gao, W.; Yoshimori, T.; Stolz, D.B.; Ron, D.; Yin, X.-M. Linking of Autophagy to Ubiquitin-Proteasome System Is Important for the Regulation of Endoplasmic Reticulum Stress and Cell Viability. Am. J. Pathol. 2007, 171, 513–524.
- Ravid, T.; Hochstrasser, M. Diversity of degradation signals in the ubiquitin–proteasome system. Nat. Rev. Mol. Cell Biol. 2008, 9, 679–689.
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.; Lichtenberg, M.; Luo, S.; et al. Regulation of Mammalian Autophagy in Physiology and Pathophysiology. Physiol. Rev. 2010, 90, 1383–1435.
- Li, J.; Chen, X.; Kang, R.; Zeh, H.; Klionsky, D.J.; Tang, D. Regulation and function of autophagy in pancreatic cancer. Autophagy 2020, 17, 3275–3296.
- Kraft, C.; Reggiori, F.; Peter, M. Selective types of autophagy in yeast. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2009, 1793, 1404–1412.
- Cuervo, A.M. Autophagy and aging: Keeping that old broom working. Trends Genet. 2008, 24, 604–612.
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473.
- Bejarano, E.; Cuervo, A.M. Chaperone-Mediated Autophagy. Proc. Am. Thorac. Soc. 2010, 7, 29–39.
- Majeski, A.E.; Fred, J. Dice, Mechanisms of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2435–2444.
- Li, W.-W.; Li, J.; Bao, J.-K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136.
- Geng, J.; Klionsky, D.J. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. EMBO Rep. 2008, 9, 859–864.
- Rubinsztein, D.C.; Shpilka, T.; Elazar, Z. Mechanisms of Autophagosome Biogenesis. Curr. Biol. 2012, 22, R29–R34.
- Mehrpour, M.; Esclatine, A.; Beau, I.; Codogno, P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010, 20, 748–762.
- Reggiori, F.; Komatsu, M.; Finley, K.; Simonsen, A. Autophagy: More Than a Nonselective Pathway. Int. J. Cell Biol. 2012, 2012, 3275–3296.
- Welter, E.; Thumm, M.; Krick, R. Quantification of nonselective bulk autophagy in S. cerevisiae using Pgk1-GFP. Autophagy 2010, 6, 794–797.
- Wang, X.; Wang, P.; Zhang, Z.; Farré, J.-C.; Li, X.; Wang, R.; Xia, Z.; Subramani, S.; Ma, C. The autophagic degradation of cytosolic pools of peroxisomal proteins by a new selective pathway. Autophagy 2020, 16, 154–166.
- Ichimura, Y.; Kumanomidou, T.; Sou, Y.-S.; Mizushima, T.; Ezaki, J.; Ueno, T.; Kominami, E.; Yamane, T.; Tanaka, K.; Komatsu, M. Structural Basis for Sorting Mechanism of p62 in Selective Autophagy. J. Biol. Chem. 2008, 283, 22847–22857.
- Itakura, E.; Mizushima, N. p62 targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J. Cell Biol. 2011, 192, 17–27.
- Kristensen, A.R.; Schandorff, S.; Høyer-Hansen, M.; Nielsen, M.O.; Jaüaüttelaü, M.; Dengjel, J.; Andersen, J.S. Ordered Organelle Degradation during Starvation-induced Autophagy. Mol. Cell. Proteom. 2008, 7, 2419–2428.
- Zhang, J.; Tripathi, D.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.-H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269.
- Burman, J.L.; Pickles, S.; Wang, C.; Sekine, S.; Vargas, J.N.S.; Zhang, Z.; Youle, A.M.; Nezich, C.L.; Wu, X.; Hammer, J.A.; et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 2017, 216, 3231–3247.
- Goldman, S.J.; Taylor, R.; Zhang, Y.; Jin, S. Autophagy and the degradation of mitochondria. Mitochondrion 2010, 10, 309–315.
- Castrejón-Jiménez, N.S.; Leyva-Paredes, K.; Hernández-González, J.C.; Luna-Herrera, J.; García-Pérez, B.E. The role of autophagy in bacterial infections. Biosci. Trends 2015, 9, 149–159.
- Benjamin, J.L.; Sumpter, R.; Levine, B.; Hooper, L.V. Intestinal Epithelial Autophagy Is Essential for Host Defense against Invasive Bacteria. Cell Host Microbe 2013, 13, 723–734.
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688.
- Luo, G.; Jian, Z.; Zhu, Y.; Chen, B.; Ma, R.; Tang, F.; Xiao, Y. Sirt1 promotes autophagy and inhibits apoptosis to protect cardiomyocytes from hypoxic stress. Int. J. Mol. Med. 2019, 43, 2033–2043.
- Mohan, N.; Banik, N.L.; Ray, S.K. Combination of N-(4-hydroxyphenyl) retinamide and apigenin suppressed starvation-induced autophagy and promoted apoptosis in malignant neuroblastoma cells. Neurosci. Lett. 2011, 502, 24–29.
- Sahni, S.; Gillson, J.; Park, K.C.; Chiang, S.; Leck, L.Y.W.; Jansson, P.; Richardson, D.R. NDRG1 suppresses basal and hypoxia-induced autophagy at both the initiation and degradation stages and sensitizes pancreatic cancer cells to lysosomal membrane permeabilization. Biochim. Biophys. Acta (BBA) Gen. Subj. 2020, 1864, 129625.
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830.
- Boya, P.; Codogno, P.; Rodriguez-Muela, N. Autophagy in stem cells: Repair, remodelling and metabolic reprogramming. Development 2018, 145, dev146506.
- Xiong, Y.; Contento, A.L.; Nguyen, P.Q.; Bassham, D.C. Degradation of Oxidized Proteins by Autophagy during Oxidative Stress in Arabidopsis. Plant Physiol. 2007, 143, 291–299.
- Hosokawa, N.; Sasaki, T.; Iemura, S.-I.; Natsume, T.; Hara, T.; Mizushima, N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009, 5, 973–979.
- Kim, J.; Guan, K.-L. Regulation of the Autophagy Initiating Kinase ULK1 By Nutrients: Roles of Mtorc1 and AMPK. Cell Cycle 2011, 10, 1337–1338.
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.-C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297.
- Martin, K.R.; Celano, S.L.; Solitro, A.R.; Gunaydin, H.; Scott, M.; O’Hagan, R.C.; Shumway, S.D.; Fuller, P.; MacKeigan, J.P. A Potent and Selective ULK1 Inhibitor Suppresses Autophagy and Sensitizes Cancer Cells to Nutrient Stress. iScience 2018, 8, 74–84.
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.-Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.-L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750.
- Di Bartolomeo, S.; Corazzari, M.; Nazio, F.; Oliverio, S.; Lisi, G.; Antonioli, M.; Pagliarini, V.; Matteoni, S.; Fuoco, C.; Giunta, L.; et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J. Cell Biol. 2010, 191, 155–168.
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 Antiapoptotic Proteins Inhibit Beclin 1-Dependent Autophagy. Cell 2005, 122, 927–939.
- Hamacher-Brady, A. Autophagy Regulation and Integration with Cell Signaling. Antioxid. Redox Signal. 2012, 17, 756–765.
- Van Humbeeck, C.; Cornelissen, T.; Vandenberghe, W. Ambra1: A Parkin-binding protein involved in mitophagy. Autophagy 2011, 7, 1555–1556.
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 Forms Two Distinct Phosphatidylinositol 3-Kinase Complexes with Mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372.
- Park, J.-M.; Seo, M.; Jung, C.H.; Grunwald, D.; Stone, M.; Otto, N.M.; Toso, E.; Ahn, Y.; Kyba, M.; Griffin, T.; et al. ULK1 phosphorylates Ser30 of BECN1 in association with ATG14 to stimulate autophagy induction. Autophagy 2018, 14, 584–597.
- Ylä-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.-L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185.
- Ge, L.; Schekman, R. The ER-Golgi intermediate compartment feeds the phagophore membrane. Autophagy 2014, 10, 170–172.
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774.
- Romanov, J.; Walczak, M.; Ibiricu, I.; Schüchner, S.; Ogris, E.; Kraft, C.; Martens, S. Mechanism and functions of membrane binding by the Atg5–Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012, 31, 4304–4317.
- Otomo, C.; Metlagel, Z.; Takaesu, G.; Otomo, T. Structure of the human ATG12~ATG5 conjugate required for LC3 lipidation in autophagy. Nat. Struct. Mol. Biol. 2013, 20, 59.
- Giménez-Xavier, P.; Francisco, R.; Platini, F.; Pérez, R.; Ambrosio, S. LC3-I conversion to LC3-II does not necessarily result in complete autophagy. Int. J. Mol. Med. 2008, 22, 781.
- Shpilka, T.; Weidberg, H.; Pietrokovski, S.; Elazar, Z. Atg8: An autophagy-related ubiquitin-like protein family. Genome Biol. 2011, 12, 226.
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2021, 17, 1–382.
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518.
- Pankiv, S.; Høyvarde Clausen, T.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145.
- Clausen, T.H.; Lamark, T.; Isakson, P.; Finley, K.D.; Larsen, K.B.; Brech, A.; Øvervatn, A.; Stenmark, H.; Bjørkøy, G.; Simonsen, A.; et al. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 2010, 6, 330–344.
- Kirkin, V.; Lamark, T.; Sou, Y.-S.; Bjørkøy, G.; Nunn, J.L.; Bruun, J.-A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A Role for NBR1 in Autophagosomal Degradation of Ubiquitinated Substrates. Mol. Cell 2009, 33, 505–516.
- Noda, N.; Ohsumi, Y.; Inagaki, F. Atg8-family interacting motif crucial for selective autophagy. FEBS Lett. 2010, 584, 1379–1385.
- Mizushima, N.; Yamamoto, A.; Hatano, M.; Kobayashi, Y.; Kabeya, Y.; Suzuki, K.; Tokuhisa, T.; Ohsumi, Y.; Yoshimori, T. Dissection of Autophagosome Formation Using Apg5-Deficient Mouse Embryonic Stem Cells. J. Cell Biol. 2001, 152, 657–668.
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455.
- Kim, Y.-M.; Jung, C.H.; Seo, M.; Kim, E.K.; Park, J.-M.; Bae, S.S.; Kim, D.-H. mTORC1 Phosphorylates UVRAG to Negatively Regulate Autophagosome and Endosome Maturation. Mol. Cell 2015, 57, 207–218.
- Liang, C.; Lee, J.-S.; Inn, K.-S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008, 10, 776–787.
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mul, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151.
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396.
- Wang, L.; Tian, Y.; Ou, J.-H.J. HCV Induces the Expression of Rubicon and UVRAG to Temporally Regulate the Maturation of Autophagosomes and Viral Replication. PLoS Pathog. 2015, 11, e1004764.
- Thomas, M.; Davis, T.; Loos, B.; Sishi, B.; Huisamen, B.; Strijdom, H.; Engelbrecht, A.-M. Autophagy is essential for the maintenance of amino acids and ATP levels during acute amino acid starvation in MDAMB231 cells. Cell Biochem. Funct. 2018, 36, 65–79.
- Onodera, J.; Ohsumi, Y. Autophagy Is Required for Maintenance of Amino Acid Levels and Protein Synthesis under Nitrogen Starvation. J. Biol. Chem. 2005, 280, 31582–31586.
- Kaushal, G.P. Autophagy protects proximal tubular cells from injury and apoptosis. Kidney Int. 2012, 82, 1250–1253.
- Wang, R.C.; Levine, B. Autophagy in cellular growth control. FEBS Lett. 2010, 584, 1417–1426.
- Sansal, I.; Sellers, W.R. The Biology and Clinical Relevance of the PTEN Tumor Suppressor Pathway. J. Clin. Oncol. 2004, 22, 2954–2963.
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2016, 109, 314–341.
- Vara, J.Á.F.; Casado, E.; De Castro, J.; Cejas, P.; Belda-Iniesta, C.; González-Barón, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204.
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71.
- Foster, K.G.; Acosta-Jaquez, H.A.; Romeo, Y.; Ekim, B.; Soliman, G.A.; Carriere, A.; Roux, P.P.; Ballif, B.A.; Fingar, D.C. Regulation of mTOR Complex 1 (mTORC1) by Raptor Ser863 and Multisite Phosphorylation. J. Biol. Chem. 2010, 285, 80–94.
- Shang, L.; Wang, X. AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy 2011, 7, 924–926.
- Linke, M.; Fritsch, S.D.; Sukhbaatar, N.; Hengstschläger, M.; Weichhart, T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett. 2017, 591, 3089–3103.
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR Pathways in Cancer and Autophagy. Cancers 2018, 10, 18.
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295.
- Galbaugh, T.; Cerrito, M.G.; Jose, C.C.; Cutler, M.L. EGF-induced activation of Akt results in mTOR-dependent p70S6 kinase phosphorylation and inhibition of HC11 cell lactogenic differentiation. BMC Cell Biol. 2006, 7, 34.
- Mollinedo, F.; Gajate, C. Novel therapeutic approaches for pancreatic cancer by combined targeting of RAF→MEK→ERK signaling and autophagy survival response. Ann. Transl. Med. 2019, 7, S153.
- Peyssonnaux, C.; Eychene, A. The Raf/MEK/ERK pathway: New concepts of activation. Biol. Cell 2001, 93, 53–62.
- Wortzel, I.; Seger, R. The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes Cancer 2011, 2, 195–209.
- Wang, J.; Whiteman, M.W.; Lian, H.; Wang, G.; Singh, A.; Huang, D.; Denmark, T. A Non-canonical MEK/ERK Signaling Pathway Regulates Autophagy via Regulating Beclin 1. J. Biol. Chem. 2009, 284, 21412–21424.
- Ugland, H.; Naderi, S.; Brech, A.; Collas, P.; Blomhoff, H.K. cAMP induces autophagy via a novel pathway involving ERK, cyclin E and Beclin 1. Autophagy 2011, 7, 1199–1211.
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.-W.; Farkas, A.M.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J.; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892.
- Tong, Y.; Huang, H.; Pan, H. Inhibition of MEK/ERK activation attenuates autophagy and potentiates pemetrexed-induced activity against HepG2 hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2015, 456, 86–91.
- Martinez-Lopez, N.; Athonvarangkul, D.; Mishall, P.; Sahu, S.; Singh, R. Autophagy proteins regulate ERK phosphorylation. Nat. Commun. 2013, 4, 2799.
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023.
- Hawley, S.A.; Ross, F.A.; Russell, F.M.; Atrih, A.; Lamont, D.J.; Hardie, D.G. Mechanism of Activation of AMPK by Cordycepin. Cell Chem. Biol. 2020, 27, 214–222.e4.
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224.
- Ross, F.A.; Jensen, T.E.; Hardie, D.G. Differential regulation by AMP and ADP of AMPK complexes containing different γ subunit isoforms. Biochem. J. 2016, 473, 189–199.
- Ke, R.; Xu, Q.; Li, C.; Luo, L.; Huang, D. Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism. Cell Biol. Int. 2018, 42, 384–392.
- Thornton, C. AMPK: Keeping the (power) house in order? Neuronal Signal. 2017, 1, NS20160020.
- Sakamoto, K.; Göransson, O.; Hardie, D.G.; Alessi, D.R. Activity of LKB1 and AMPK-related kinases in skeletal muscle: Effects of contraction, phenformin, and AICAR. Am. J. Physiol. Metab. 2004, 287, E310–E317.
- Neumann, D. Is TAK1 a direct upstream kinase of AMPK? Int. J. Mol. Sci. 2018, 19, 2412.
- Peng, R.; Chen, Y.; Wei, L.; Li, G. Resistance to FGFR1-targeted therapy leads to autophagy via TAK1/AMPK activation in gastric cancer. Gastric Cancer 2020, 23, 988–1002.
- Dunlop, E.A.; Tee, A. The Kinase Triad, AMPK, mTORC1 and ULK1, Maintains Energy and Nutrient Homoeostasis. Biochem. Soc. Trans. 2013, 41, 939–943.
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80.
- Pattingre, S. The Antiapoptotic Protein BCL-2 Has Also an Antiautophagy Role Through Beclin 1 Inhibition. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Elsevier: Amsterdam, The Netherlands, 2016; Volume 9, pp. 165–174.
- Xu, H.-D.; Qin, Z.-H. Beclin 1, Bcl-2 and autophagy. Autophagy Biol. Dis. 2019, 1206, 109–126.
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-Mediated Phosphorylation of Bcl-2 Regulates Starvation-Induced Autophagy. Mol. Cell 2008, 30, 678–688.
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580.
- Yang, S.; Kimmelman, A.C. A critical role for autophagy in pancreatic cancer. Autophagy 2011, 7, 912–913.
- Endo, S.; Nakata, K.; Ohuchida, K.; Takesue, S.; Nakayama, H.; Abe, T.; Koikawa, K.; Okumura, T.; Sada, M.; Horioka, K.; et al. Autophagy Is Required for Activation of Pancreatic Stellate Cells, Associated with Pancreatic Cancer Progression and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 2017, 152, 1492–1506.e24.
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729.
- Grasso, D.; Garcia, M.N.; Iovanna, J.L. Autophagy in pancreatic cancer. Int. J. Cell Biol. 2012, 2012, 760498.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Farrow, B.; Albo, D.; Berger, D.H. The Role of the Tumor Microenvironment in the Progression of Pancreatic Cancer. J. Surg. Res. 2008, 149, 319–328.
- Hu, H.-F.; Ye, Z.; Qin, Y.; Xu, X.-W.; Yu, X.-J.; Zhuo, Q.-F.; Ji, S.-R. Mutations in key driver genes of pancreatic cancer: Molecularly targeted therapies and other clinical implications. Acta Pharmacol. Sin. 2021, 42, 1725–1741.
- Kopp, J.L.; Dubois, C.L.; Schaeffer, D.F.; Samani, A.; Taghizadeh, F.; Cowan, R.W.; Rhim, A.D.; Stiles, B.L.; Valasek, M.; Sander, M. Loss of Pten and Activation of Kras Synergistically Induce Formation of Intraductal Papillary Mucinous Neoplasia from Pancreatic Ductal Cells in Mice. Gastroenterology 2018, 154, 1509–1523.e5.
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100.
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640.
- Ciccarese, F.; Zulato, E.; Indraccolo, S. LKB1/AMPK pathway and drug response in cancer: A therapeutic perspective. Oxidative Med. Cell. Longev. 2019, 2019, 8730816.
- Zadra, G.; Batista, J.L.; Loda, M. Dissecting the Dual Role of AMPK in Cancer: From Experimental to Human Studies. Mol. Cancer Res. 2015, 13, 1059–1072.
- Eichner, L.J.; Brun, S.N.; Herzig, S.; Young, N.P.; Curtis, S.D.; Shackelford, D.B.; Shokhirev, M.N.; Leblanc, M.; Vera, L.I.; Hutchins, A.; et al. Genetic Analysis Reveals AMPK Is Required to Support Tumor Growth in Murine Kras-Dependent Lung Cancer Models. Cell Metab. 2019, 29, 285–302.e7.
- Jin, X.; Dai, L.; Ma, Y.; Wang, J.; Liu, Z. Implications of HIF-1α in the tumorigenesis and progression of pancreatic cancer. Cancer Cell Int. 2020, 20, 273.
- Zhao, H.; Wu, S.; Li, H.; Duan, Q.; Zhang, Z.; Shen, Q.; Wang, C.; Yin, T. ROS/KRAS/AMPK Signaling Contributes to Gemcitabine-Induced Stem-like Cell Properties in Pancreatic Cancer. Mol. Ther. Oncolytics 2019, 14, 299–312.
- Storz, P. Oxidative Stress in Cancer. In Oxidative Stress and Redox Regulation; Springer: Berlin/Heidelberg, Germany, 2013; pp. 427–447.
- Cadet, J.; Wagner, J.R. DNA Base Damage by Reactive Oxygen Species, Oxidizing Agents, and UV Radiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012559.
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38.
- Chavez-Dominguez, R.; Perez-Medina, M.; Lopez-Gonzalez, J.S.; Galicia-Velasco, M.; Aguilar-Cazares, D. The Double-Edge Sword of Autophagy in Cancer: From Tumor Suppression to Pro-tumor Activity. Front. Oncol. 2020, 10, 2064.
- Duffy, J.P.; Eibl, G.; Reber, H.A.; Hines, O.J. Influence of hypoxia and neoangiogenesis on the growth of pancreatic cancer. Mol. Cancer 2003, 2, 12.
- Delrue, L.; Blanckaert, P.; Mertens, D.; Cesmeli, E.; Ceelen, W.P.; Duyck, P. Assessment of Tumor Vascularization in Pancreatic Adenocarcinoma Using 128-Slice Perfusion Computed Tomography Imaging. J. Comput. Assist. Tomogr. 2011, 35, 434–438.
- Kong, B.; Cheng, T.; Wu, W.; Regel, I.; Raulefs, S.; Friess, H.; Erkan, M.; Esposito, I.; Kleeff, J.; Michalski, C.W. Hypoxia-induced endoplasmic reticulum stress characterizes a necrotic phenotype of pancreatic cancer. Oncotarget 2015, 6, 32154–32160.
- Kobayashi, H.; Watanabe, R.; Choyke, P.L. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics 2014, 4, 81.
- Zhu, J.; Chen, Y.; Jin, Y.; Yu, Y.; Zhang, X.; Zhou, J. Gemcitabine induces apoptosis and autophagy via the AMPK/mTOR signaling pathway in pancreatic cancer cells. Biotechnol. Appl. Biochem. 2018, 65, 665–671.
- Zeh, H.J.; Bahary, N.; Boone, B.A.; Singhi, A.D.; Miller-Ocuin, J.L.; Normolle, D.P.; Zureikat, A.H.; Hogg, M.E.; Bartlett, D.L.; Lee, K.K.; et al. A Randomized Phase II Preoperative Study of Autophagy Inhibition with High-Dose Hydroxychloroquine and Gemcitabine/Nab-Paclitaxel in Pancreatic Cancer Patients. Clin. Cancer Res. 2020, 26, 3126–3134.
- Chen, X.; Tao, Y.; He, M.; Deng, M.; Guo, R.; Sheng, Q.; Wang, X.; Ren, K.; Li, T.; He, X. Co-delivery of autophagy inhibitor and gemcitabine using a pH-activatable core-shell nanobomb inhibits pancreatic cancer progression and metastasis. Theranostics 2021, 11, 8692.
- Mathiassen, S.G.; De Zio, D.; Cecconi, F. Autophagy and the Cell Cycle: A Complex Landscape. Front. Oncol. 2017, 7, 51.
- Jiang, S.-H.; Li, J.; Dong, F.-Y.; Yang, J.-Y.; Liu, D.-J.; Yang, X.-M.; Wang, Y.-H.; Yang, M.-W.; Fu, X.-L.; Zhang, X.-X.; et al. Increased Serotonin Signaling Contributes to the Warburg Effect in Pancreatic Tumor Cells Under Metabolic Stress and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 2017, 153, 277–291.e19.
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218.
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 2012, 149, 656–670.
- Seo, J.-W.; Choi, J.; Lee, S.-Y.; Sung, S.; Yoo, H.J.; Kang, M.-J.; Cheong, H.; Son, J. Autophagy is required for PDAC glutamine metabolism. Sci. Rep. 2016, 6, 37594.
- Vonlaufen, A.; Phillips, P.A.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Pancreatic Stellate Cells and Pancreatic Cancer Cells: An Unholy Alliance. Cancer Res. 2008, 68, 7707–7710.
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483.
- Nguyen, A.V.; Trompetto, B.; Tan, X.H.M.; Scott, M.B.; Hu, K.H.-H.; Deeds, E.; Butte, M.J.; Chiou, P.Y.; Rowat, A.C. Differential Contributions of Actin and Myosin to the Physical Phenotypes and Invasion of Pancreatic Cancer Cells. Cell. Mol. Bioeng. 2020, 13, 27–44.
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy Promotes Immune Evasion of Pancreatic Cancer by Degrading MHC-I. Nature 2020, 581, 100–105.
- Zhang, W.; He, R.; Yanh, W.; Zhang, Y.; Yuan, Q.; Wang, J.; Liu, Y.; Chen, S.; Zhang, S.; Zhang, W.; et al. Autophagic Schwann cells promote perineural invasion mediated by the NGF/ATG7 paracrine pathway in pancreatic cancer. J. Exp. Clin. Cancer Res. 2022, 41, 48.
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28.
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027.
- Sohal, D.P.S.; Kennedy, E.B.; Khorana, A.; Copur, M.S.; Crane, C.H.; Garrido-Laguna, I.; Krishnamurthi, S.; Moravek, C.; O’Reilly, E.M.; Philip, P.A.; et al. Metastatic Pancreatic Cancer: ASCO Clinical Practice Guideline Update. J. Clin. Oncol. 2018, 36, 2545–2556.
- Hosein, A.N.; Dougan, S.K.; Aguirre, A.J.; Maitra, A. Translational advances in pancreatic ductal adenocarcinoma therapy. Nat. Cancer 2022, 3, 272–286.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cancer. Br. J. Cancer 2020, 124, 333–344.
- Folkerts, H.; Hilgendorf, S.; Vellenga, E.; Bremer, E.; Wiersma, V.R. The multifaceted role of autophagy in cancer and the microenvironment. Med. Res. Rev. 2019, 39, 517–560.
- Dower, C.M.; Wills, C.A.; Frisch, S.M.; Wang, H.-G. Mechanisms and context underlying the role of autophagy in cancer metastasis. Autophagy 2018, 14, 1110–1128.
- Turner, C.E. Paxillin and focal adhesion signalling. Nat. Cell Biol. 2000, 2, E231–E236.
- Wang, Y.; Yin, S.; Zhang, L.; Shi, K.; Tang, J.; Zhang, Z.; He, Q. A tumor-activatable particle with antimetastatic potential in breast cancer via inhibiting the autophagy-dependent disassembly of focal adhesion. Biomaterials 2018, 168, 1–9.
- Sharifi, M.N.; Mowers, E.E.; Drake, L.E.; Collier, C.; Chen, H.; Zamora, M.; Mui, S.; Macleod, K.F. Autophagy Promotes Focal Adhesion Disassembly and Cell Motility of Metastatic Tumor Cells through the Direct Interaction of Paxillin with LC3. Cell Rep. 2016, 15, 1660–1672.
- Zhu, H.; Wang, D.; Zhang, L.; Xie, X.; Wu, Y.; Liu, Y.; Shao, G.; Su, Z. Upregulation of autophagy by hypoxia-inducible factor-1α promotes EMT and metastatic ability of CD133+ pancreatic cancer stem-like cells during intermittent hypoxia. Oncol. Rep. 2014, 32, 935–942.
- Sahni, S.; Bae, D.-H.; Lane, D.; Kovacevic, Z.; Kalinowski, D.S.; Jansson, P.J.; Richardson, D.R. The Metastasis Suppressor, N-myc Downstream-regulated Gene 1 (NDRG1), Inhibits Stress-induced Autophagy in Cancer Cells. J. Biol. Chem. 2014, 289, 9692–9709.
- Merlot, A.; Porter, G.; Sahni, S.; Lim, E.; Peres, P.; Richardson, D. The metastasis suppressor, NDRG1, differentially modulates the endoplasmic reticulum stress response. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 2094–2110.
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435.
- Philipson, E.; Engström, C.; Naredi, P.; Bourghardt Fagman, J. High expression of p62/SQSTM1 predicts shorter survival for patients with pancreatic cancer. BMC Cancer 2022, 22, 347.
- Todoric, J.; Antonucci, L.; Di Caro, G.; Li, N.; Wu, X.; Lytle, N.K.; Dhar, D.; Banerjee, S.; Fagman, J.B.; Browne, C.D.; et al. Stress-Activated NRF2-MDM2 Cascade Controls Neoplastic Progression in Pancreas. Cancer Cell 2017, 32, 824–839.e8.
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.-F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. KrasG12D-induced IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 105–120.
- Nomura, A.; Majumder, K.; Giri, B.; Dauer, P.; Dudeja, V.; Roy, S.; Banerjee, S.; Saluja, A.K. Inhibition of NF-kappa B pathway leads to deregulation of epithelial–mesenchymal transition and neural invasion in pancreatic cancer. Lab. Investig. 2016, 96, 1268–1278.
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. NF-kB in development and progression of human cancer. Virchows Arch. 2005, 446, 475–482.
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323.
- Sergeant, G.; Vankelecom, H.; Gremeaux, L.; Topal, B. Role of cancer stem cells in pancreatic ductal adenocarcinoma. Nat. Rev. Clin. Oncol. 2009, 6, 580–586.
- Rausch, V.; Liu, L.; Apel, A.; Rettig, T.; Gladkich, J.; Labsch, S.; Kallifatidis, G.; Kaczorowski, A.; Groth, A.; Gross, W.; et al. Autophagy mediates survival of pancreatic tumour-initiating cells in a hypoxic microenvironment. J. Pathol. 2012, 227, 325–335.
- Kizaka-Kondoh, S.; Itasaka, S.; Zeng, L.; Tanaka, S.; Zhao, T.; Takahashi, Y.; Shibuya, K.; Hirota, K.; Semenza, G.L.; Hiraoka, M. Selective Killing of Hypoxia-Inducible Factor-1–Active Cells Improves Survival in a Mouse Model of Invasive and Metastatic Pancreatic Cancer. Clin. Cancer Res. 2009, 15, 3433–3441.
- Yang, M.-C.; Wang, H.-C.; Hou, Y.-C.; Tung, H.-L.; Chiu, T.-J.; Shan, Y.-S. Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol. Cancer 2015, 14, 179.
- Akar, U.; Ozpolat, B.; Mehta, K.; Fok, J.; Kondo, Y.; Lopez-Berestein, G. Tissue Transglutaminase Inhibits Autophagy in Pancreatic Cancer Cells. Mol. Cancer Res. 2007, 5, 241–249.
- Görgülü, K.; Diakopoulos, K.; Ai, J.; Schoeps, B.; Kabacaoglu, D.; Karpathaki, A.-F.; Ciecielski, K.J.; Aksoy, E.K.; Ruess, D.A.; Berninger, A.; et al. Levels of the Autophagy-Related 5 Protein Affect Progression and Metastasis of Pancreatic Tumors in Mice. Gastroenterology 2019, 156, 203–217.e20.
More