Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Poater Albert and Version 2 by Camila Xu.
The essential constituents of a UV curing system are a resin, which is an oligomer whose backbone confers the properties to the final polymer; a monomer, which acts as a cross-linking agent and adjusts the viscosity of the mixture to an acceptable level for application; and a photoinitiator, which is responsible for the light absorbance and governs the curing depth and rate.
- coating
- cross-linking
- photopolymerization
- curing
1. Introduction
The essential constituents of a UV curing system are a resin [1][62], which is an oligomer whose backbone confers the properties to the final polymer; a monomer, which acts as a cross-linking agent and adjusts the viscosity of the mixture to an acceptable level for application; and a photoinitiator [2][63], which is responsible for the light absorbance and governs the curing depth and rate [3][4][20,41]. All of them participate in the cross-linked free radical polymerization reaction and are incorporated into the final polymer.
2. Resins
A resin is an oligomer, which is a chain formed by the union of monomer units, that will constitute the framework of the cured polymer network [5][6][55,64]. It cannot be considered a polymer because the latter is a macromolecule with a much larger number of monomer units [7][65], whereas these oligomers usually contain from 1 to 12 repetitive units [8][66]. They are usually formed through step-growth polymerization, a type of polymerization mechanism in which bifunctional or multifunctional monomers react to form first dimers, then trimers and, eventually, long-chain oligomers [9][67]. The type of monomer and their length, together with the cure extension, will determine the properties of the final polymer [10][11][38,68].
The main classes of UV-curable resins that can be polymerized by a radical mechanism are unsaturated and acrylate resins [3][20]. The most common backbone structure for unsaturated resins is polyesters, and for acrylate resins, they are polyurethanes, although other structures, such as polyesters, can also be used [12][13][69,70].
2.1. Unsaturated Resins
Unsaturated resins periodically contain monomers with double bonds in their backbone, which will react during the free radical polymerization. They are generally polyesters, which means that they have ester linkages in their backbone chain, generated through condensation reactions between diols and unsaturated dicarboxylic acids, also called esterification reactions (Scheme 1Scheme 7) [14][71]. More than one type of each reagent could be used, obtaining then an oligomer with over three different monomers. In any case, the unsaturations come from the structure of the diacid.
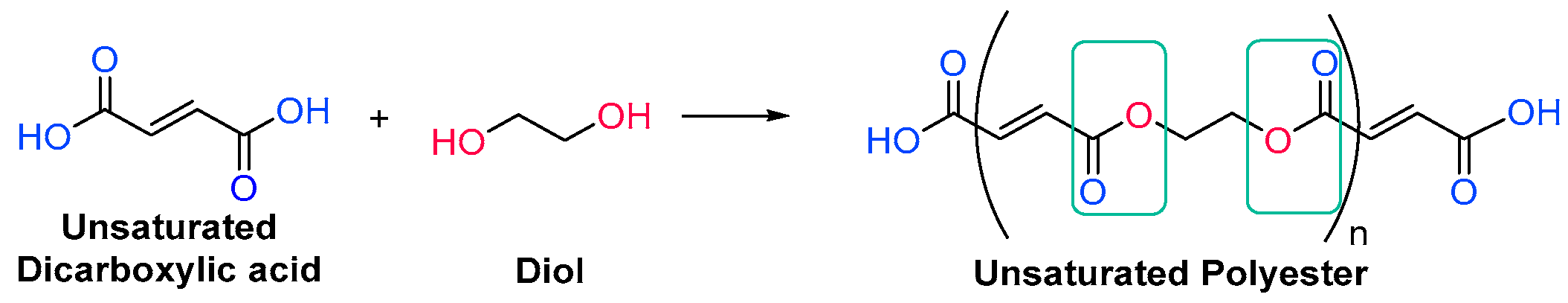
Scheme 17. Generic synthesis of an unsaturated polyester oligomer.
Generic synthesis of an unsaturated polyester oligomer.
The Fischer esterification specifically refers to the acid-catalyzed reaction of carboxylic acids and alcohols. It is one of the methods that can be employed to synthesize polyester oligomers [15][16][72,73]. The alcohol from the diol nucleophilically attacks a protonated dicarboxylic acid, and after proton transfer, a water molecule is lost from the structure of the diacid. The resulting product is in an ester, which, since both reactants are difunctionalized, still contains an alcohol and a carboxylic acid group, able to further condensate and create a chain of esters (Scheme 2Scheme 8).
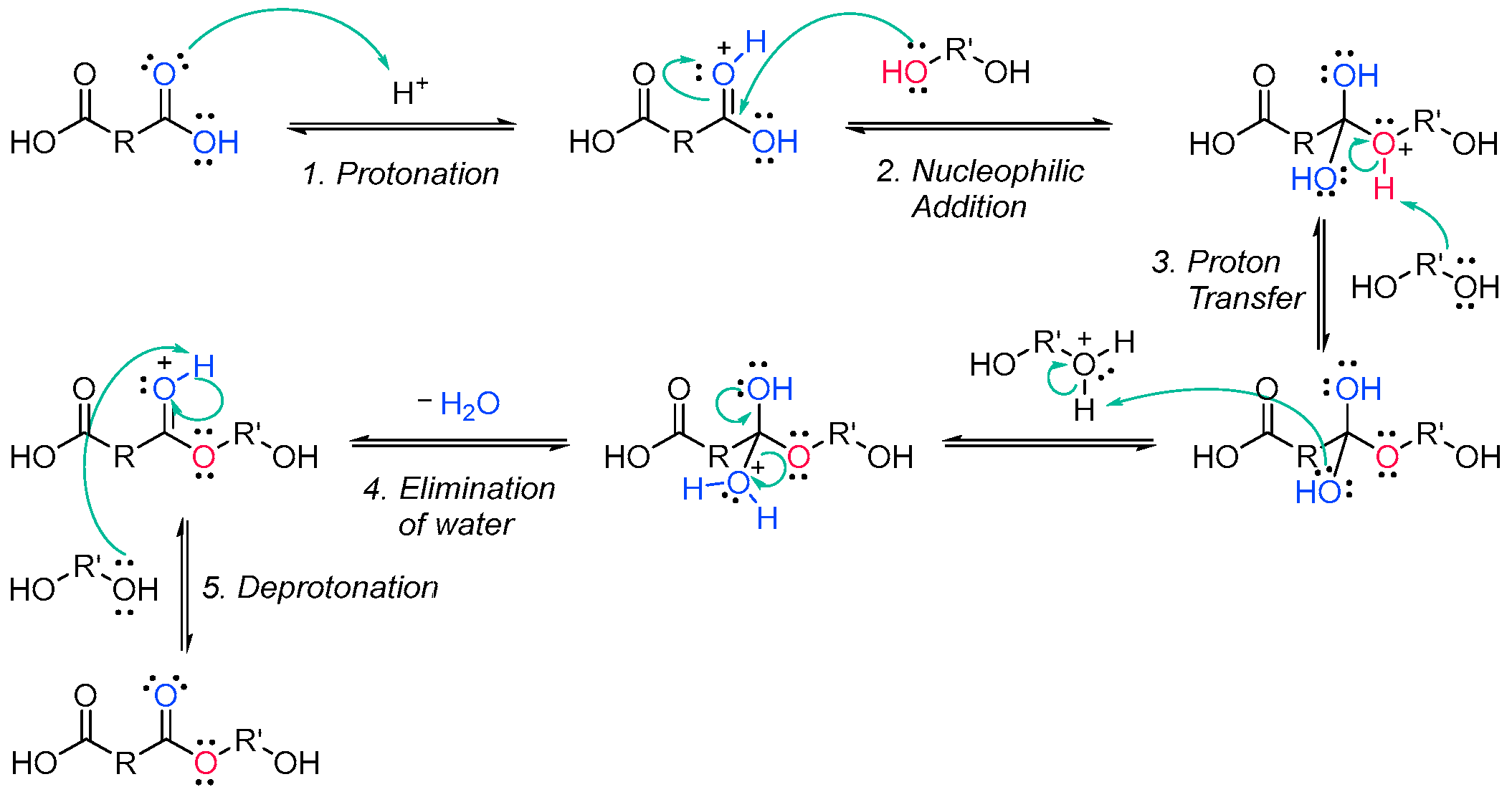
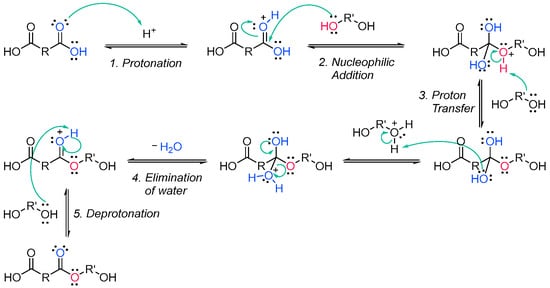
2.2. Acrylate Resins
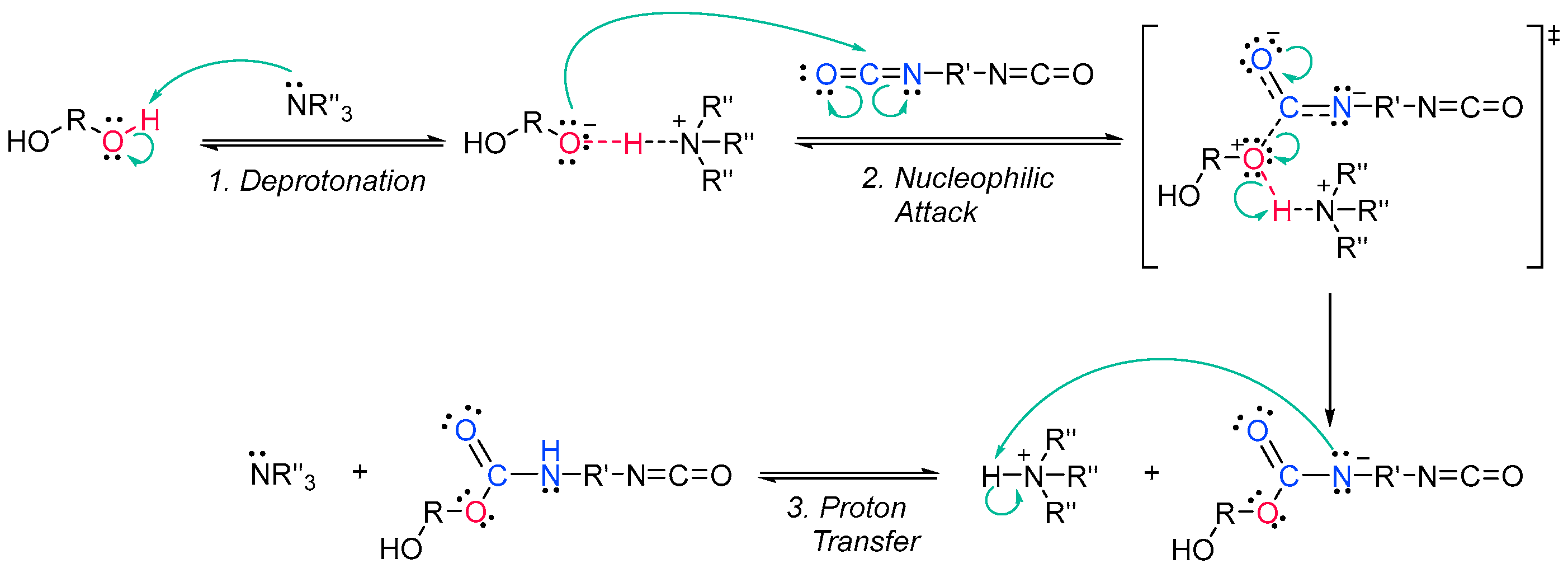
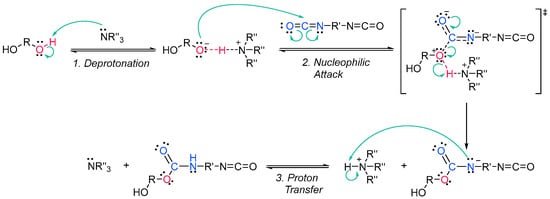
Acrylate resins contain acrylate or methacrylate groups at their ends that will react during the free radical polymerization [17][74]. They are more efficient than unsaturated resins for UV curing. Urethane acrylates, which contain a polyurethane backbone, are the most common, but polyester or polyether backbones are also used (Scheme 3Scheme 9) [18][43]. Given that all acrylates are derived from oil, which is a scarce material and also highly polluting, environmental pressure from climate change forces reusearchers to reduce the use of oil and/or look for alternative solutions, such as the conversion of renewable biomass into materials, polymers and composites [19][75]. The development and application of bio-based materials is therefore aimed at replacing commercial UV-curable acrylate resins. In detail, they are mainly epoxy, polyurethane and polyether acrylate oligomers. Resins have different functions depending on the chain structure. The evolution of the polymer chain structure has made polyester acrylate oligomers increasingly functional. Commercially, this has been replicated in the growing UV curing market. However, there are also drawbacks, caused by the fact that products of relatively low intensity are needed. Despite the disadvantage of high viscosity, polyester acrylates (PEA) lead to UV-curable resins with good hardness, high tear resistance and wear resistance, ozone resistance and polarity [20][76]. Looking for ways to reduce viscosity, and being part of the spectrum of non-petroleum-derived biodegradable polymers [21][22][77,78], poly(lactic acid) (PLA) and the use of biocurable UV coatings offer green advantages to the industry [23][24][79,80], PLA still has weaknesses although it is applied prematurely, and is far from the market. In particular, its resistance needs to be improved, especially at high temperatures. In order to influence improvements, in contrast to the linear polymers [25][26][27][81,82,83], the networking of PLA and the incorporation of star-shaped chains have been described, as well as modification by copolymerization with poly(ε-caprolactone) (PCL) to improve its hardness, also favoring the positive reduction of viscosity [28][84]. Notably, recently, maleimides have appeared to be competitors of acrylates in photopolymerization because they can operate without a photoinitiator and also because their polymerization rate is directly competitive with that of acrylates [29][30][85,86]. Nonetheless, polyurethanes are synthesized similarly to polyesters; however, the polyol reacts with a di- or triisocyanate instead of a diacid [31][32][87,88]. The polyol does not necessarily have to be a polyether; for instance, polyesters could also be used [33][89]. The synthesis is catalyzed by a tertiary amine, which, according to Farka’s mechanism, interacts with a proton source to form a complex that subsequently reacts with the isocyanate (Scheme 4Scheme 10) [34][90].
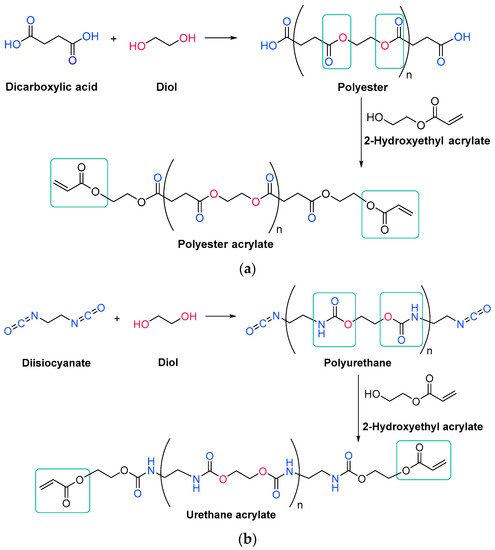
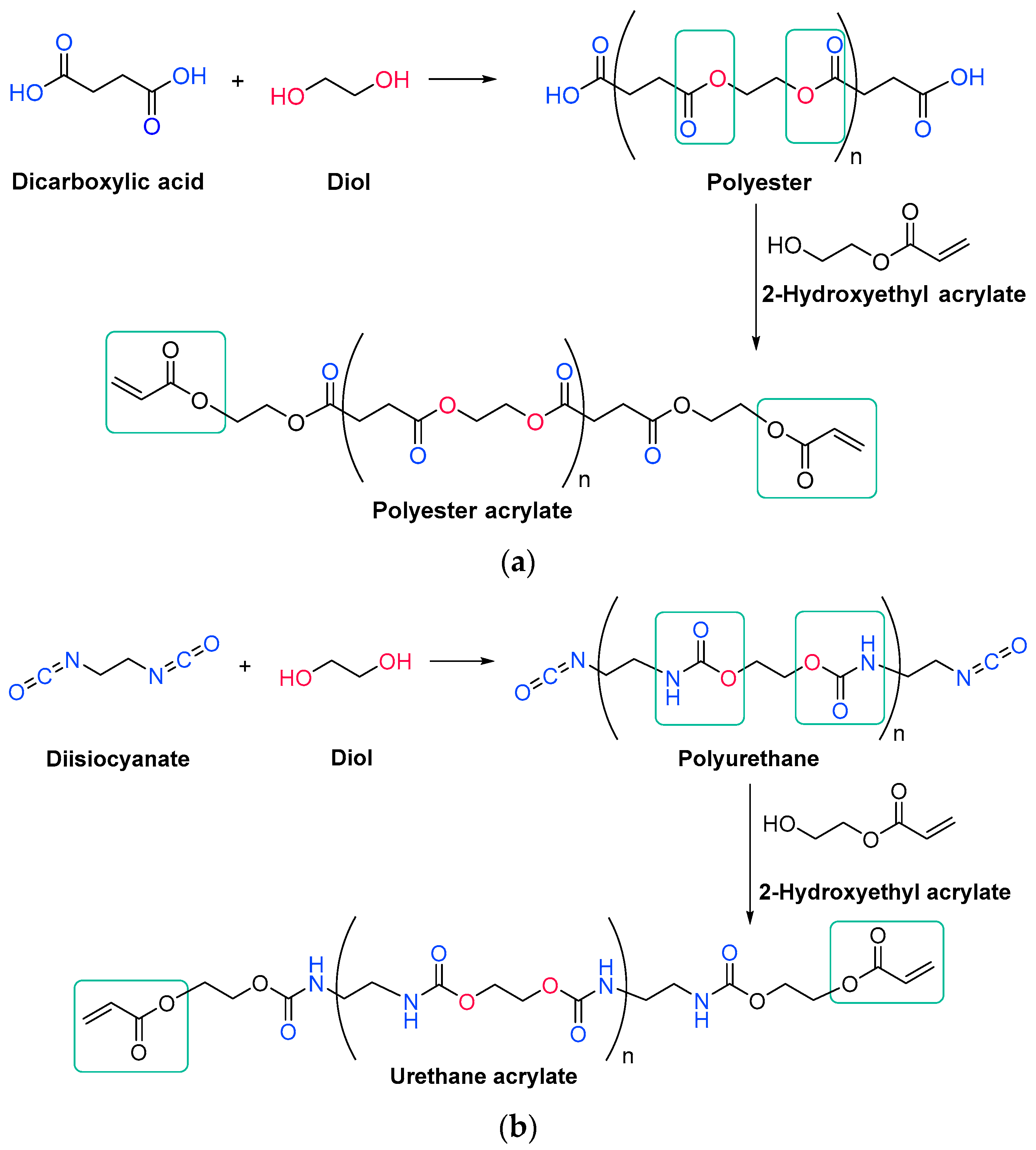
Scheme 39.
Generic synthesis of (
a
) a polyester acrylate oligomer and (
The urethane acrylate oligomer can be divided into soft and hard sections. The soft segments are produced from the polyols, and the longer they are, the more flexible is the resin. The hard segments are produced from isocyanate and are immobile and stiff; moreover, they can form intermolecular hydrogen bonds between the hydrogen attached to the nitrogen and the carbonyl oxygen [33][36][89,95].
3. Monomers
Monofunctional monomers are used to lower the viscosity of the mixture and to add flexibility to the final polymer thanks to their cross-linking ability. In order to incorporate into the polymer, they must contain carbon–carbon double bonds [18][43]. Amongst the most common monofunctional monomers are unsaturated monomers, often used with unsaturated polyester resins, acrylate monomers [37][96], and usually paired with acrylate resins and thiol monomers, which can be added as curing accelerators [38][39][97,98].
3.1. Unsaturated Monomers
The unsaturated monomer per excellence is styrene, a small molecule with little steric hindrance in comparison to the resin. Scheme 5 Scheme 11 depicts how styrene forms cross-linked chains between unsaturated oligomers.
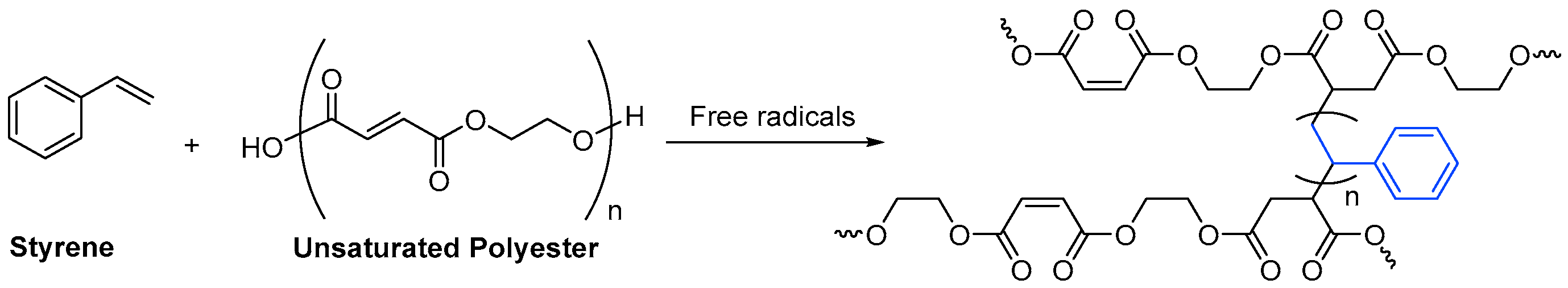
Scheme 511. Formation of cross-links by the reaction of styrene with an unsaturated polyester oligomer.
Formation of cross-links by the reaction of styrene with an unsaturated polyester oligomer.
3.2. Acrylate Monomers
A great variety of acrylate monomers are available, so that the ideal monomer or combination of monomers can be chosen in order to adjust the flexibility of the final polymer (Scheme 6Scheme 12).
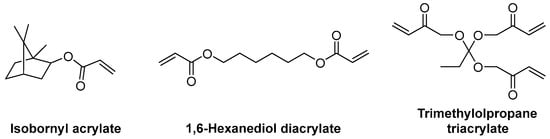
The number of unsaturated groups will affect the flexibility of the final polymer. A higher number of unsaturations will form a higher number of cross-links and will cause the polymer to be more rigid. On the other hand, flexibility can be increased by using either long-chain linear monomers such as 1,6-hexanediol diacrylate, which will link oligomers while still allowing them to move, or bulky monomers with high steric effects, such as isobornyl acrylate, which will hinder the formation of cross-links near them [38][97].
A special type of acrylate monomer is phosphates (Scheme 7Scheme 13a). They act as adherence promotors to metal surfaces, making them useful in certain applications [40][99]. Phosphate anions can replace hydroxy anions on metal oxide surfaces, so that the phosphate is adsorbed into the metal surface, making its removal difficult since chemical bonds have been formed [41][100].
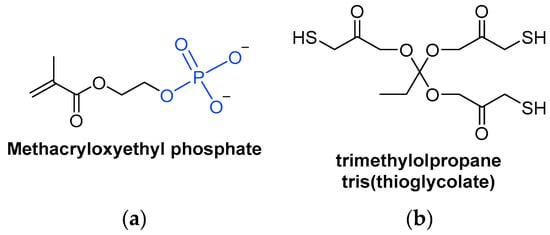
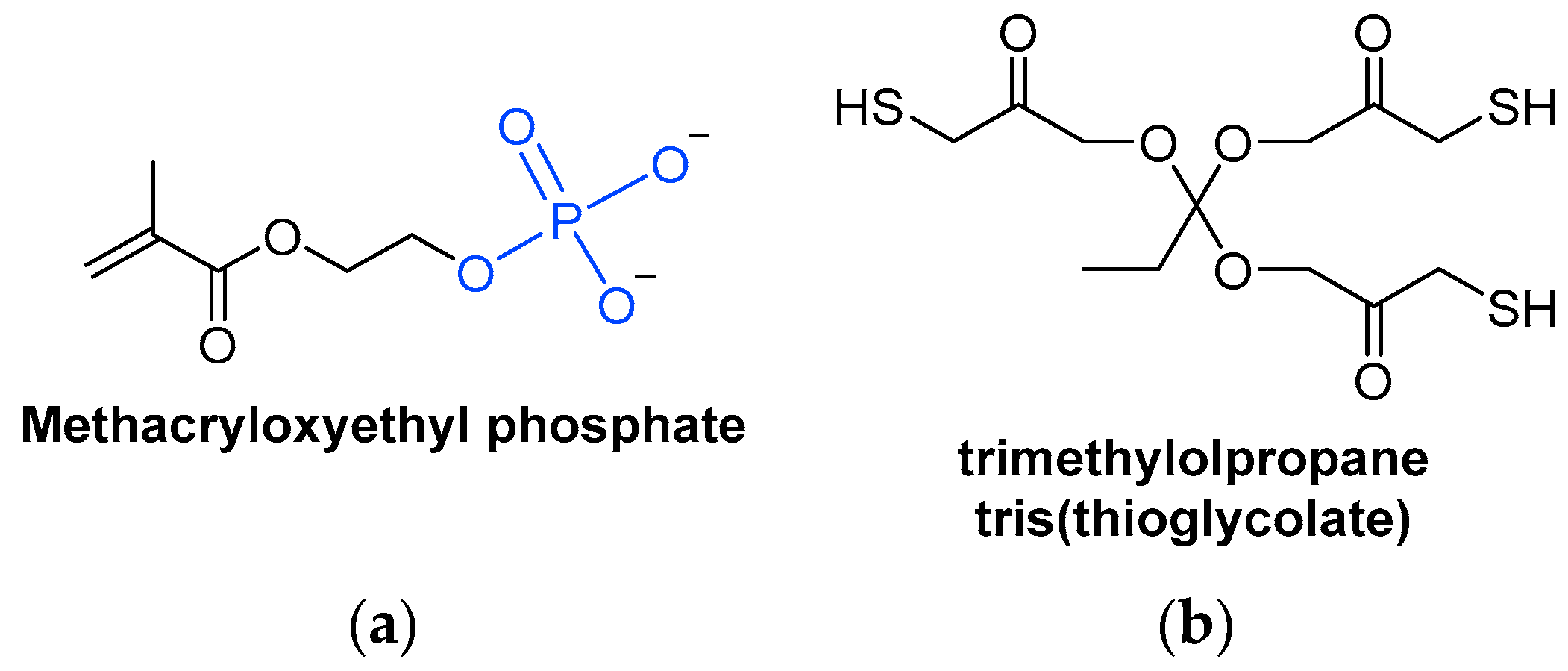
Scheme 713.
Chemical structures of (
a
) a methacrylate phosphate monomer and (
b) a trifunctionalized thiol monomer.
) a trifunctionalized thiol monomer.
3.3. Thiol Monomers
Thiol monomers combined with ene monomers can form thiol-ene systems, which are suitable UV-curable resins (Scheme 7Scheme 13b). However, monomers are more volatile than oligomers. Hence, thiols are rarely used as monomers due to their unpleasant odor, but can be used as oligomers, which are less volatile [18][43].
Their main advantage is that little to no photoinitiator is required in order to polymerize since thiols can function both as monomers and photoinitiators [42][101]. When exposed to UV light, they produce a thiyl and a hydrogen radical pair through sulfur–hydrogen bond cleavage [18][43]. However, this process is not as efficient as with an initiator; for this reason, the initiating species is often generated from the hydrogen abstraction reaction between a photoinitiator and the thiol (Scheme 8Scheme 14) [43][44][102,103]. The resulting thiyl radical adds to a double bond of a monomer and, from here, the rest of the mechanism proceeds in the same way as in Scheme 9Scheme 1. Given that most of the thiols used are polyfunctionalized, they act as powerful cross-linking agents [45][46][56,104].
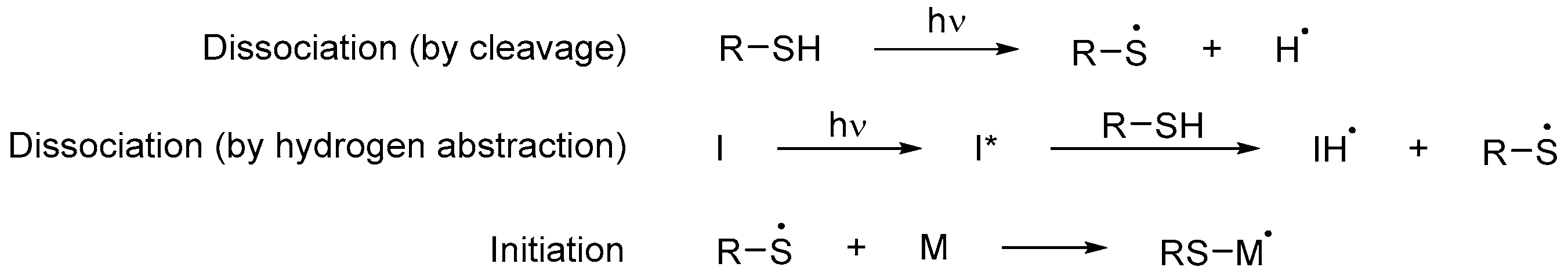
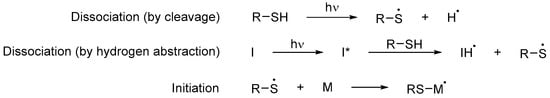
Scheme 814. Initiation step of a free radical polymerization using thiols as both photoinitiators and monomers, where I is a photoinitiator and M any monomer, including those that are part of an oligomer [45][56].
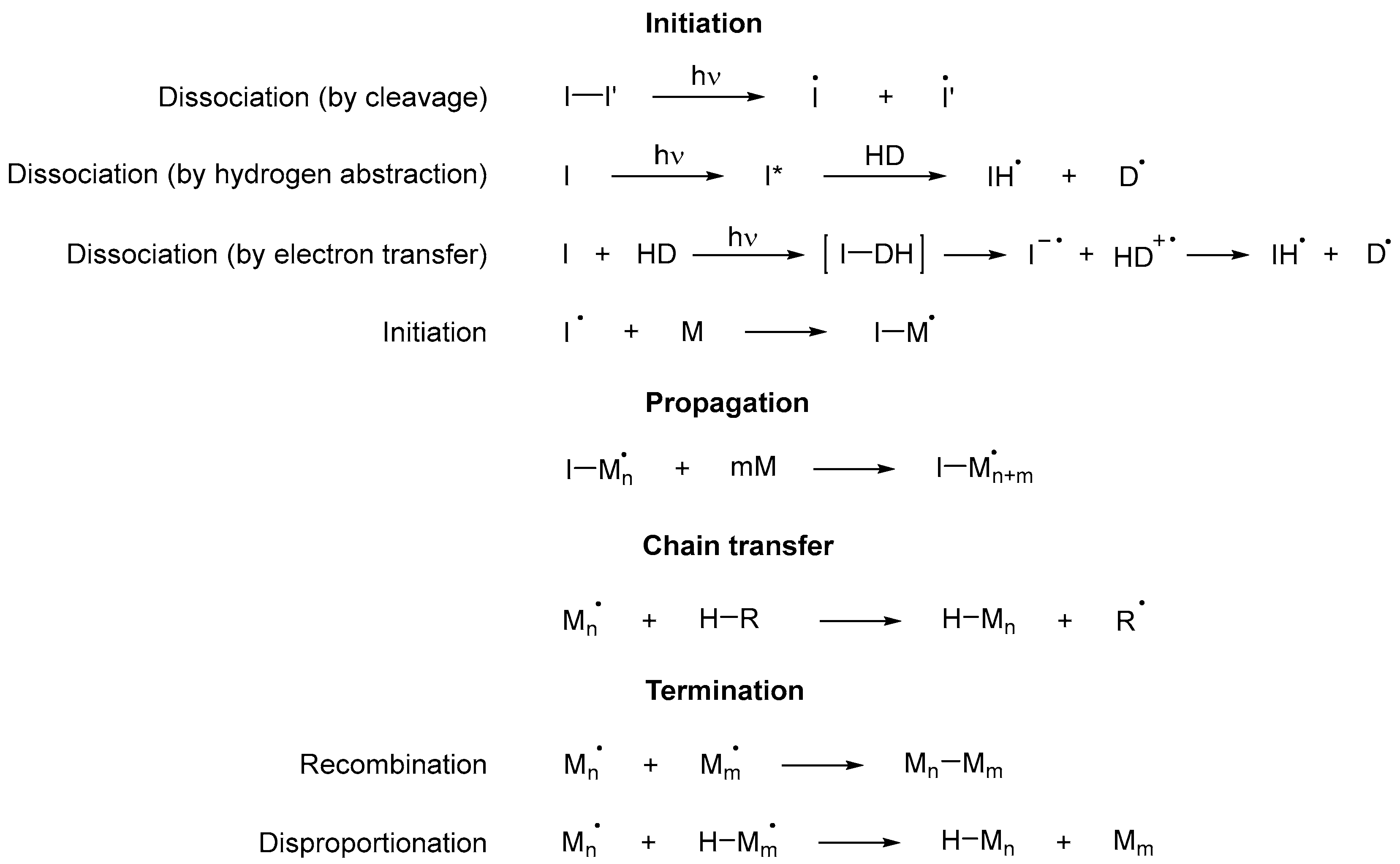 Scheme 9. Steps of a cross-linked free radical photopolymerization reaction, where I is a photoinitiator, D a donor and M any monomer, including those that are part of an oligomer [43].
Scheme 9. Steps of a cross-linked free radical photopolymerization reaction, where I is a photoinitiator, D a donor and M any monomer, including those that are part of an oligomer [43].4. Photoinitiators
Photoinitiators are able to convert light energy into chemical energy in the form of a reactive species, which can be radicals or cations, leading to the initiation of the polymerization chain. In the case of free radical polymerization reactions, radical photoinitiators are used. They are considered essential components of UV curing systems because most of the commonly used monomers are not able to generate free radicals upon exposure to UV light [3][20].
4.1. Unimolecular Photoinitiators
Photoinitiator systems termed unimolecular involve only one molecular species to generate the radical active species through homolytic cleavage. These photoinitiators are typically acetophenone derivatives, benzoin ethers, amino ketones or phosphine oxide derivatives. In most cases, the cleavage may occur in the α-position to the carbonyl group (Norrish Type I), but it can occur at the α-position in the presence weak bonds such as carbon–halogen, carbon–nitrogen, carbon–oxygen or carbon–sulfur next to the carbonyl moiety (Scheme 10Scheme 15). One of the products of α-cleavage is always a benzoyl radical, while in β-cleavage, it is always a phenacyl radical. The other radical formed will depend on the structure of the initial photoinitiator, and it will not always be active—it could often be disproportionate or recombine [18][43].
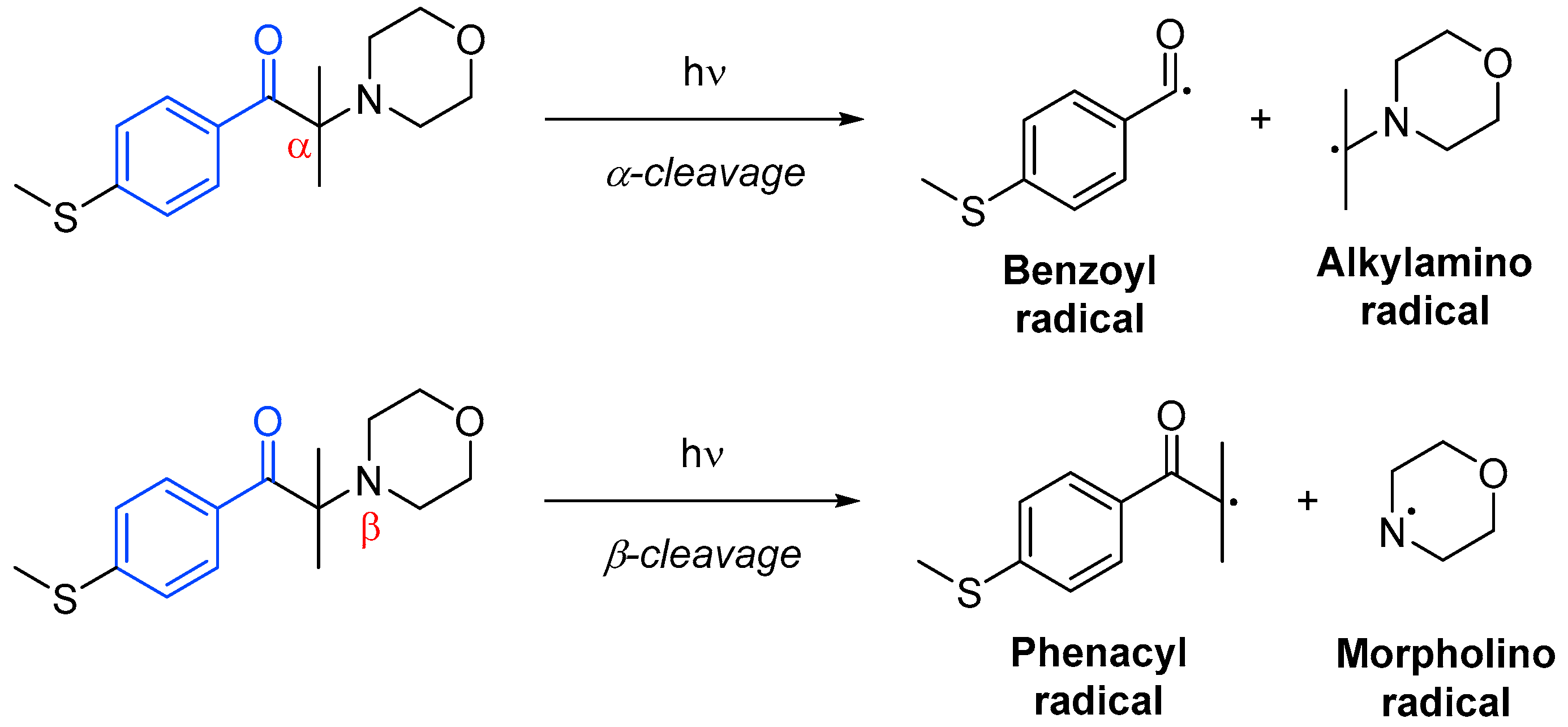
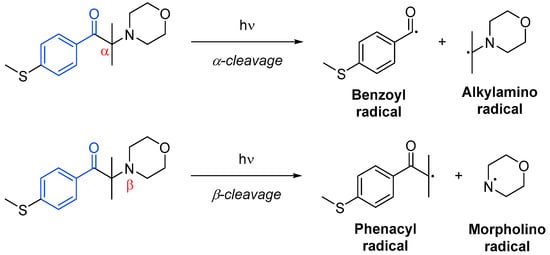
Other types of unimolecular photoinitiators are those that can form biradicals through intramolecular hydrogen abstraction (Norrish Type II). This occurs in molecules with a hydrogen atom in the α-position, able to undergo an intramolecular [49][50][1,5]-hydrogen shift. The resulting ketyl radical will most likely terminate by coupling with another free radical species, while the other radical will initiate polymerization [18][51][43,46].
Upon the absorption of light with a specific frequency, photoinitiators are promoted from the ground electronic state to an excited singlet state, from where they can undergo inter-system crossing to a triplet state of comparable energy (Figure 13). It is from the triplet state that the molecule will cleave, generating the radical species [3][52][20,107]. However, Figure 13, supplemented with the cleavage reaction of triplets only, could be somewhat incomplete since the unimolecular photoinitiators may cleave either from the singlet or triplet state, depending on the photoinitiator structure, but the cleavage of singlets is more likely due to their higher energy. Bimolecular photoinitiators work usually from the triplet state, because the lifetime of most of singlet states is too short to enable bimolecular reactions.
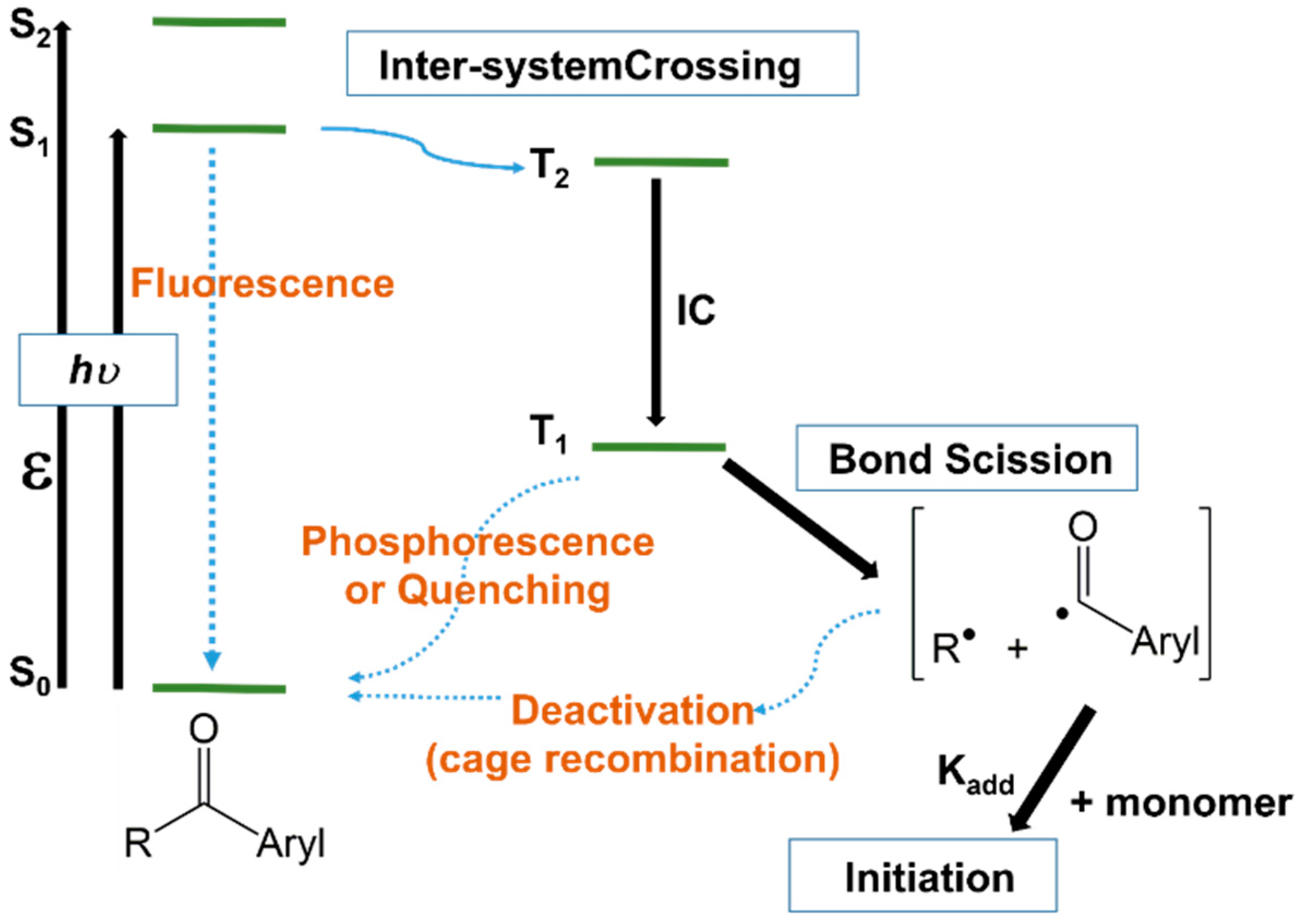
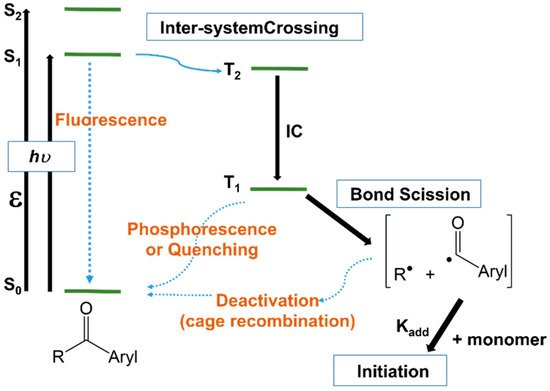
Figure 13. Jablonski diagram illustrating the photoinitiation of a type I acetophenone initiator.
Jablonski diagram illustrating the photoinitiation of a type I acetophenone initiator.
4.2. Bimolecular Photoinitiators
Photoinitiator systems termed bimolecular involve a photoinitiator that absorbs light, and a co-initiator that serves as a hydrogen or electron donor. In both cases, the formation of radicals takes place when the photoinitiator is either in the singlet or triplet excited states. These photoinitiators are typically benzophenone derivatives, thioxanthones, camphorquinones, benzyls or ketocoumarins [18][43].
In initiation by hydrogen abstraction, the co-initiator is usually an ether or an alcohol with an α-hydrogen. The resulting ether or alcohol radical will be the only initiating species. On the other hand, in photoinitiation by electron transfer, the co-initiator is typically an amine, and it forms an excited-state complex with the photoinitiator, from where electron transfer occurs. It is immediately followed by the proton transfer of an α-hydrogen from the amine, resulting in an active amine radical capable of initiating polymerization. The fact that an amine is present in the system helps to neutralize oxygen inhibition. In both cases, a ketyl radical is formed, which only participates in termination (Scheme 11Scheme 16) [18][51][53][43,46,108].
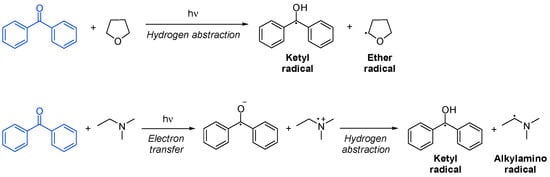
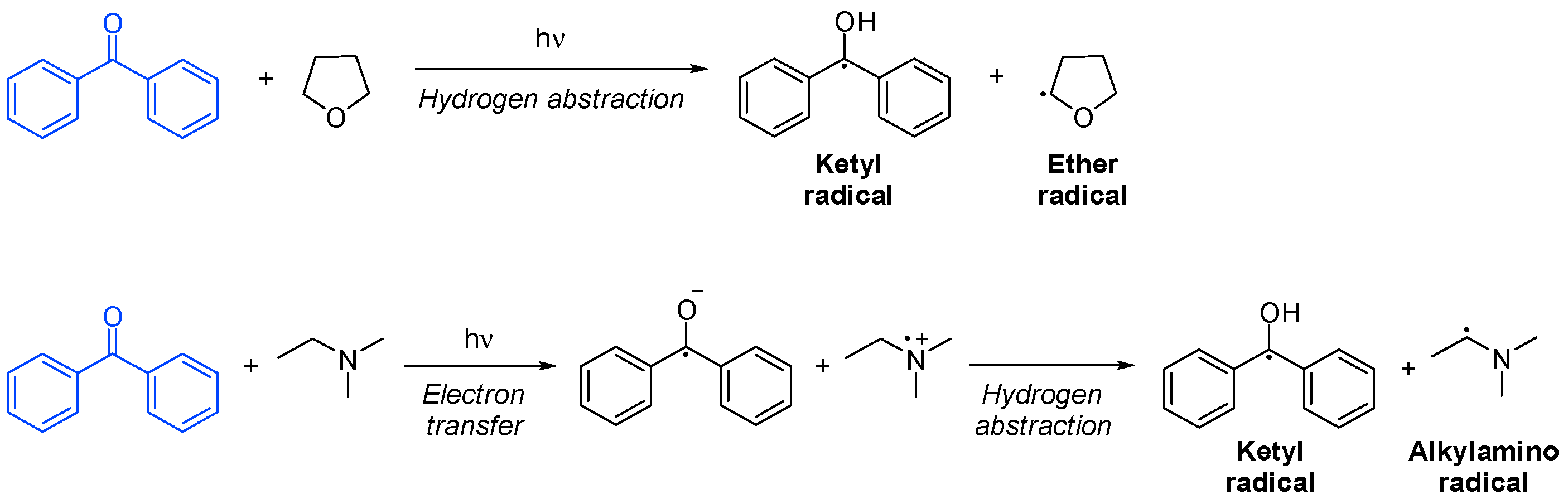
Scheme 116. Hydrogen abstraction and electron transfer of a photoinitiator based on benzophenone [53].
In addition, there are other bimolecular photoinitiators, not described here, which are among the most common components used in photoinitiating systems in the last decade, including, among others, polymethine dyes [54][55][52,109], squaric acid derivatives [56][57][58][110,111,112] or BODIPY dyes [58][59][112,113].
5. UV Light
UV light could be considered the fourth essential component of UV curing. In physics, the term ‘light’ refers to electromagnetic radiation of any wavelength. The spectrum of electromagnetic radiation can be organized by decreasing wavelength and thus increasing energy into radio waves, microwaves, IR radiation, visible light, UV radiation, X-rays and gamma rays (Figure 24). Radiation within the UV spectrum can be further divided by wavelength into UVA (315–400 nm), UVB (280–315 nm) and UVC (100–280 nm) [60][114]. The sun emits mainly visible light and infrared radiation, but it also emits some UV radiation. Of the UV light that reaches the Earth’s surface, more than 95% is UVA, with a small remainder of UVB and almost no UVC [61][115].
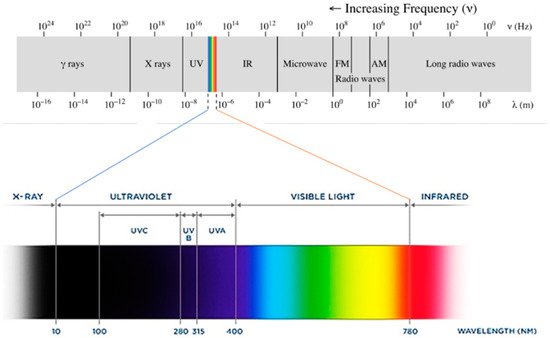
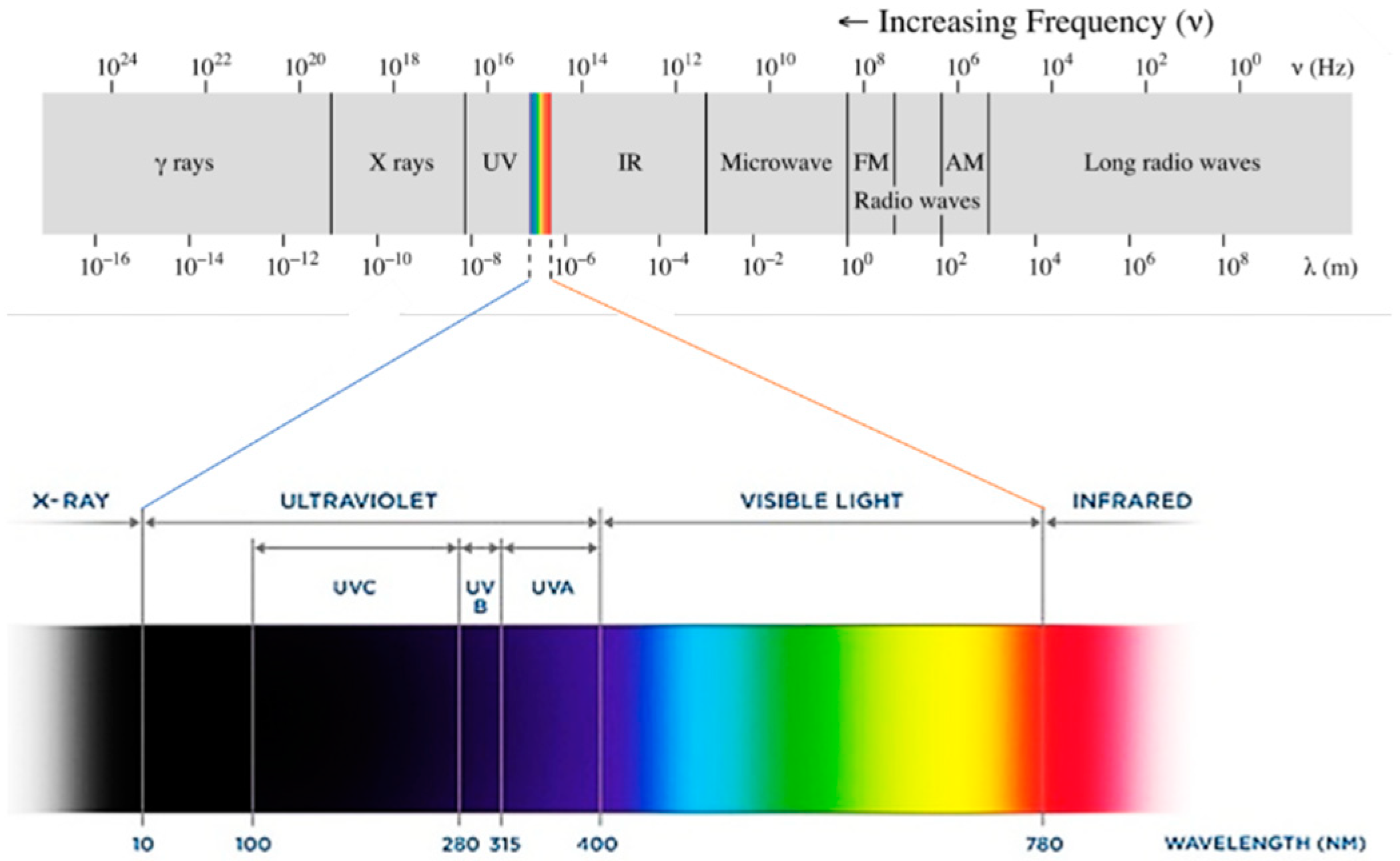
Figure 24. Electromagnetic radiation spectrum.
Electromagnetic radiation spectrum.
Each type of radiation will interact differently with matter depending on how energetic it is. Microwaves only cause changes in the rotational states of atoms and molecules, IR radiation can also trigger vibrational transitions [62][116], visible and UV light are energetic enough to modify the electronic structure by exciting outer-shell electrons, and X-rays can excite inner-shell electrons (Figure 35).
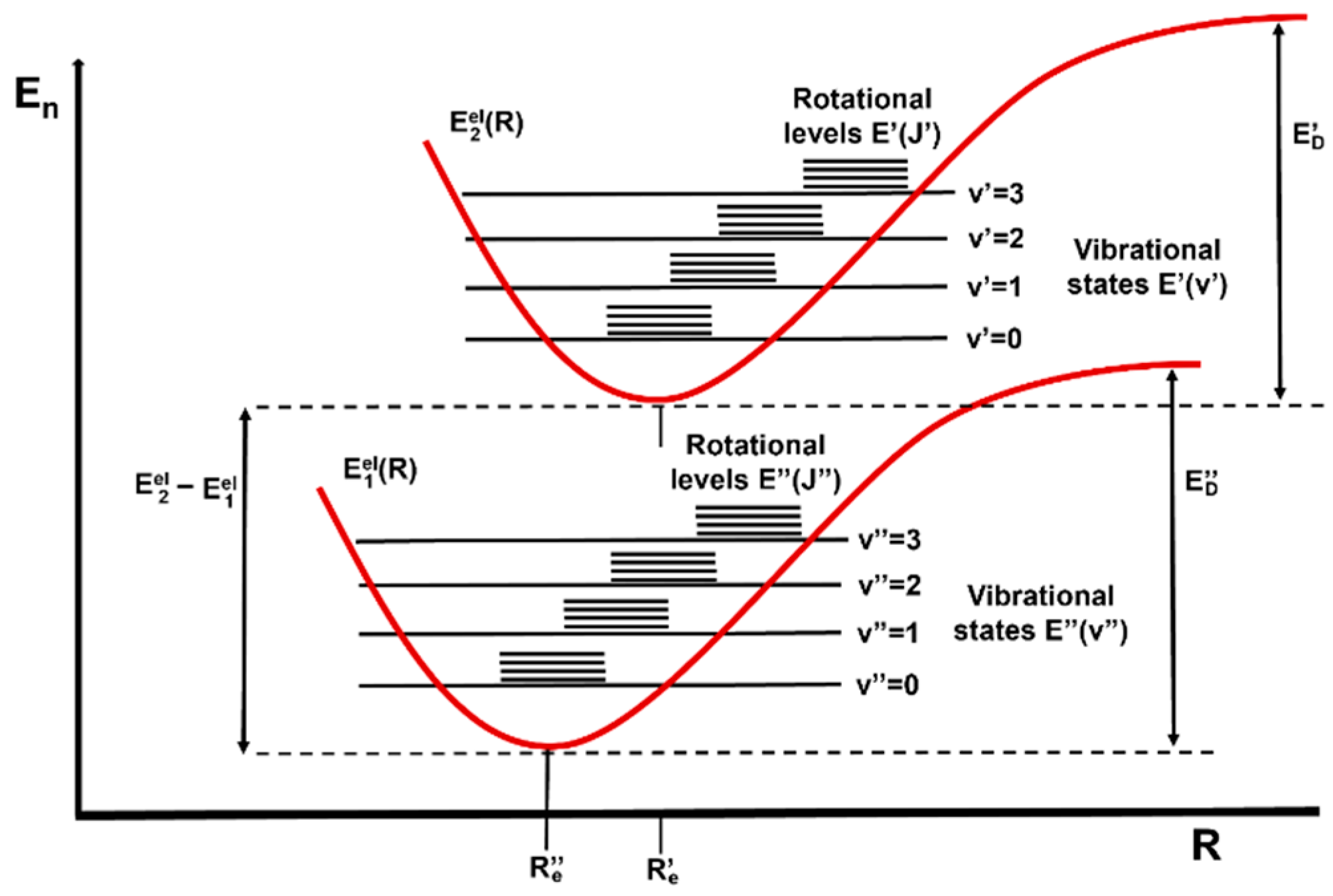
Figure 35.
Schematic representation of electronic (E
el), vibrational (E(v)) and rotational (E(J)) ground and excited states of a diatomic molecule.
), vibrational (E(v)) and rotational (E(J)) ground and excited states of a diatomic molecule.
The fact that UV and visible light can excite valence shell electrons means that it can trigger chemical reactions such as free radical photopolymerization. In organic molecules containing σ, π and n electrons, the absorption of UV–vis radiation is restricted to those molecules that contain chromophore functional groups with valence electrons of low excitation energy, such as photoinitiators. The electronic transitions that may occur in these systems are depicted in Figure 46. However, among the outlined transitions, only n → π* and π → π*, the two lowest in energy, are available in the UV–vis spectrum [63][64][117,118].
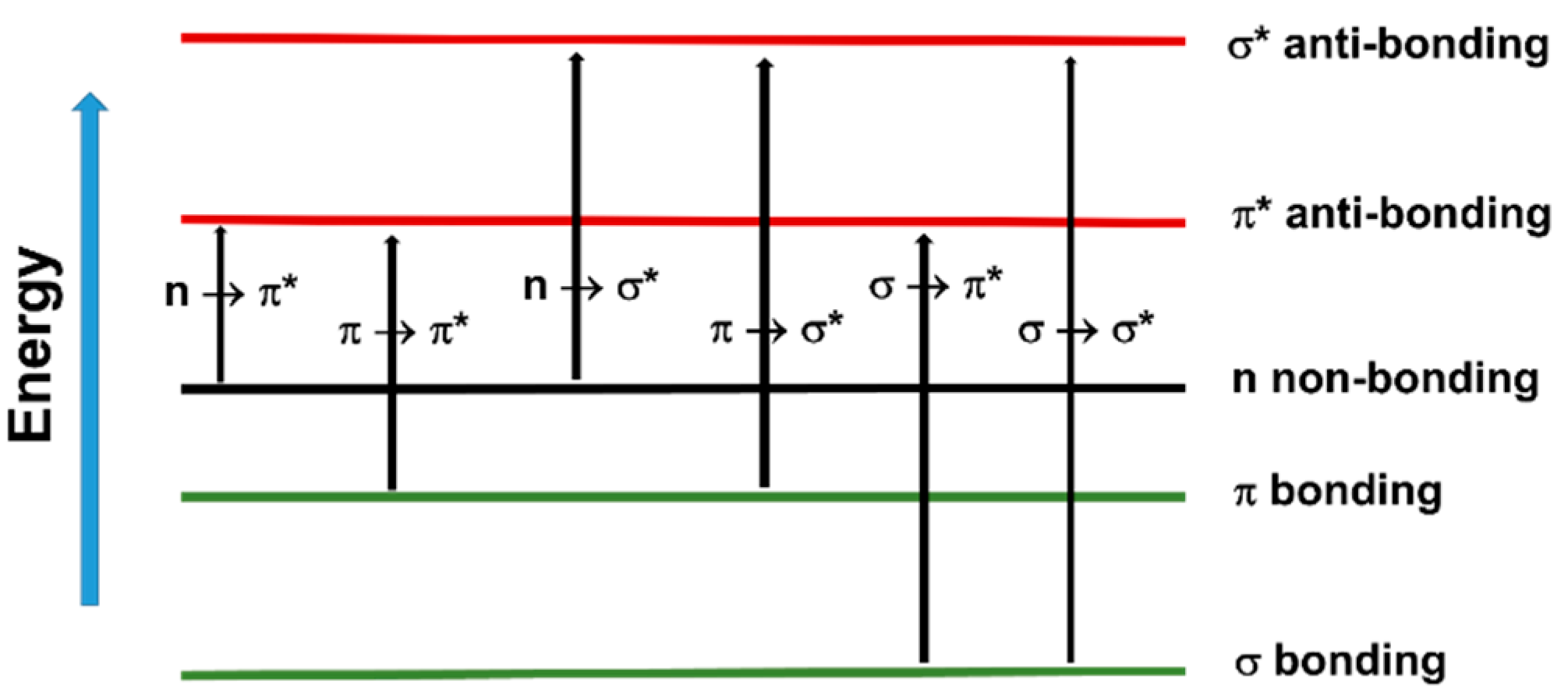
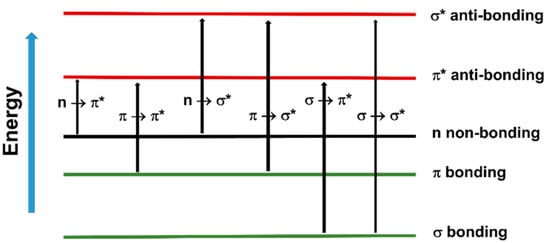
Figure 46. Electron transitions that may occur in organic molecules.
Electron transitions that may occur in organic molecules.
For the UV curing to be viable, the absorption spectrum of the photoinitiator must overlap with the emission spectrum of the light source [18][43]. The most common types of UV lamps are mercury and Light Emitting Diode (LED) lamps. The irradiation wavelength for mercury lamps ranges from 185 to 650 nm; however, they are being substituted by the less hazardous LED lamps, which irradiate at a much narrower range of 390–400 nm, the least energetic UV radiation. However, the recent developments in LED technology, emitting at 365–370 nm, have allowed the design of novel, powerful and efficient light sources that lead to the free radical and cationic photopolymerization of monomers [65][119], up to the synthesis of interpenetrating polymer networks (IPNs) [66][120].
Figure 57 depicts the absorption spectrum of some unimolecular and bimolecular photoinitiators [67][121]. It can be seen how photoinitiators A and C absorb at the UV LED wavelength range, while photoinitiators B and C would not be activated with an LED lamp.
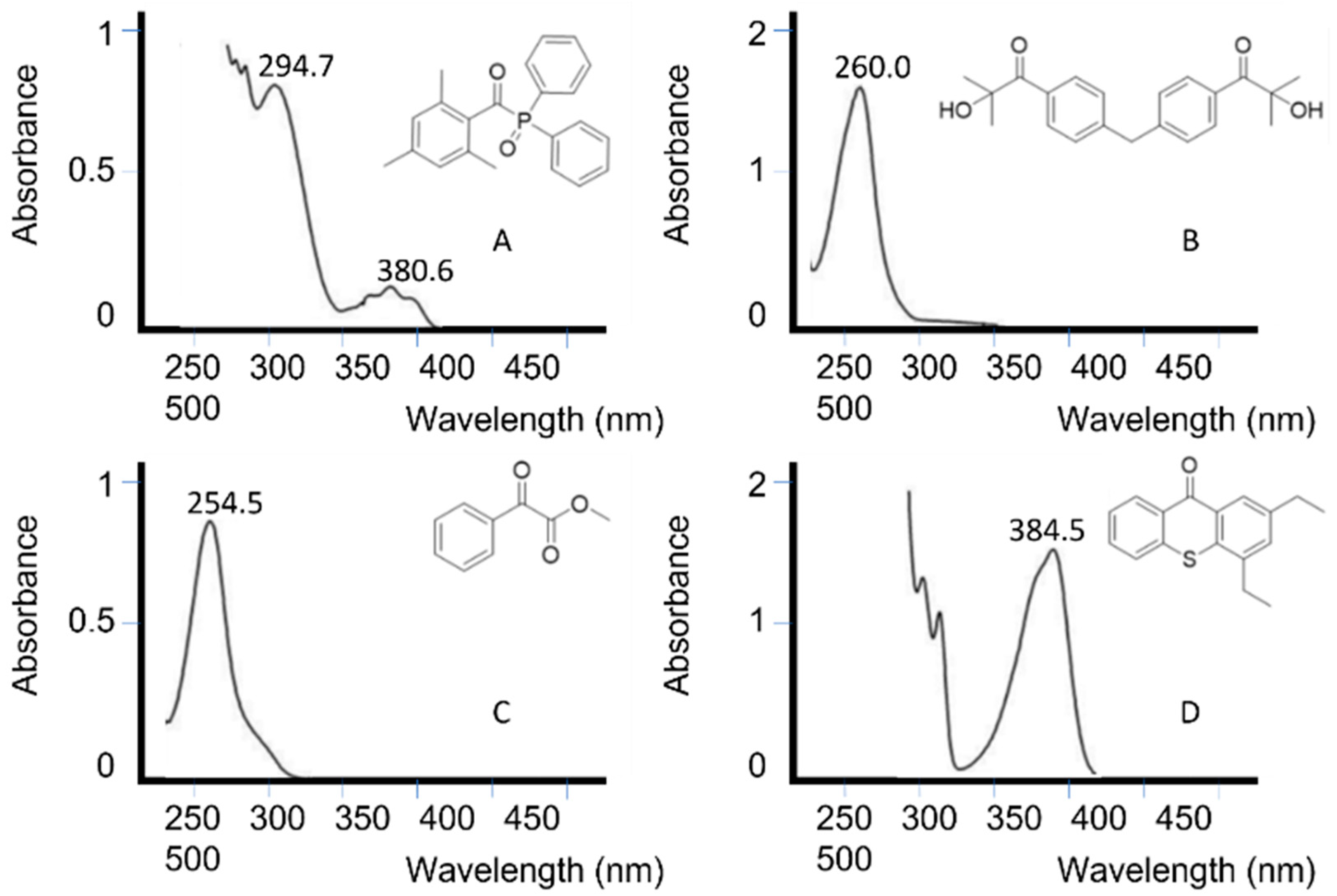
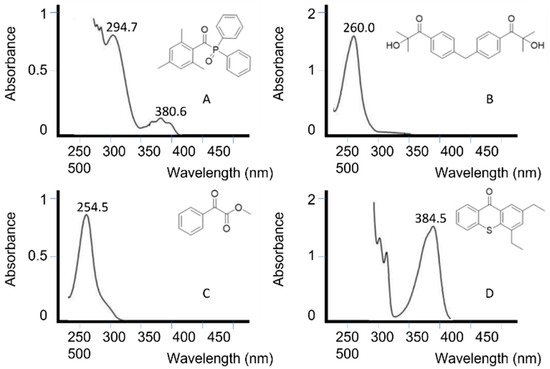
The overlap between the initiator and light source must preferably not coincide with the absorption peaks of other components in the photopolymerization [68][122], such as monomers or pigments. In systems where there is overlap, higher light intensities and photoinitiator concentrations are often used.