vVaccines have played a significant role in protecting public and personal health against infectious diseases and proved their great potential in battling cancers as well.
- therapeutic vaccine
- tumor antigens
- adjuvants
- delivery systems
- combination therapies
1. Introduction
2. Tumor Antigens for Cancer Vaccines
The principles of vaccine development, however, eluded the grasp of researchers in the context of cancer immunotherapy until 1957, when Prehn and Main showed that an immune response could be induced in mice against carcinogen-induced sarcomas, which also prevented mice from developing tumors when further challenged with the same tumor cells [6]. This was further solidified in 1991 when van der Burrgen and colleagues discovered the tumor antigen encoding gene MZ2-E through complimentary DNA (cDNA) transfections in cells with relevant major histocompatibility complexes (MHC). After this, relevant transfectants were able to be identified by anti-tumor cytotoxic T-lymphocytes (CTLs) [7]. Since then, a great number of advances have been made in the discovery of novel tumor antigens. MHCs are the key components of the adaptive immune system that recognize foreign proteins. They are expressed on the surface of most nucleated non-immune and immune cells and present antigenic peptide fragments to either CD8+ or CD4+ T-cells for an adaptive immune response [8,9,10][8][9][10]. The general structure of an MHC is composed of immunoglobulin-like anchoring peptides, which fixes the MHC to the exterior of cellular membrane and a peptide-binding region (PBR), which is responsible for antigen recognition and presentation to the T-cell receptors (TCR). MHC molecules are further subcategorized into class I and class II molecules [8,9,10,11,12][8][9][10][11][12]. MHC class I molecules are heterodimeric molecules that are made up of two polypeptide chains: an α chain that is comprised of three domains (α1, α2, α3) and a smaller β2-microglobulin chain [13]. The α1 and α2 domains are key components of the PBR on MHC I and their inherent polymorphisms mitigate and influence antigenic peptides binding to the PBR for antigen presentation to CD8+ CTLs [9,12,13][9][12][13]. MHC class I molecules are consistently expressed on the surfaces of most nucleated cells except for sperm cells and select neuronal cells. MHC class II molecules are also heterodimeric molecules that are composed of an α chain and a β chain; however, they have two distinct domains referred to as α1, α2 and β1, β2 [8,12][8][12]. The PBR in MHC II is composed of α1 and β1 domains [8,10,12][8][10][12]. MHC class II molecules are only expressed on antigen presenting cells (APCs) such as dendritic cells (DCs), macrophages, and B-cells, which specifically present to CD4+ T cells for an immune response [10,12,14][10][12][14]. If a tumor antigen was presented restrictively on an MHC class I molecule to an appropriately matched TCR on a CD8+ CTL, then cytolytic actions will be carried out by CTLs, leading to the shrinkage or elimination of the tumor and an established immunity against the same tumor antigens [15,16,17][15][16][17]. Cytolytic activity is also seen when tumor antigens are presented restrictively on MHC class II molecules for tandem antigen recognition between CD4+ T-cell TCR and MHC class II molecule [18,19,20][18][19][20]. However, this is not the main anti-tumor effect enacted by CD4+ T-cells but a more assistive role observed via the activation of immune effector pathways that stimulates cytokine production and other aspects of the innate immune system [21,22][21][22].2.1. Classification of Tumor Antigens
Tumor antigens were classified into two categories: tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs) (Figure 1). TAAs are tumor antigens that are expressed in normal germline cells as well as tumor cells [23]. Due to the wide expression profile of TAAs, they have been further subcategorized into differentiation tumor antigens and overexpressed tumor antigens [23]. Overexpressed tumor antigens are a class of TAAs that could be found in normal tissues but are expressed at an elevated level in various cancerous tissues [23,24][23][24]. A well-known example is human epidermal growth factor receptor 2 (also known as HER2 or ERBB2). Differentiation tumor antigens are a class of TAAs that are limited in their expression to one tissue type and show lineage-specific expression. These antigens are expressed in tumors and normal cells that are derived from the same cellular origin [24,25][24][25]. Antigens derived from melanocyte differentiation proteins are typical differentiation antigens that are expressed in melanomas but can be expressed in normal skin melanocytes and retinal tissue [25,26][25][26]. Due to the “self” nature of these antigens, there will be a propensity for the development of autoimmune disorders, such as the occurrence of vitiligo after the chemoimmunotherapy of metastatic melanoma [27]. More examples of TAAs and TSAs were summarized in the supplementary tables.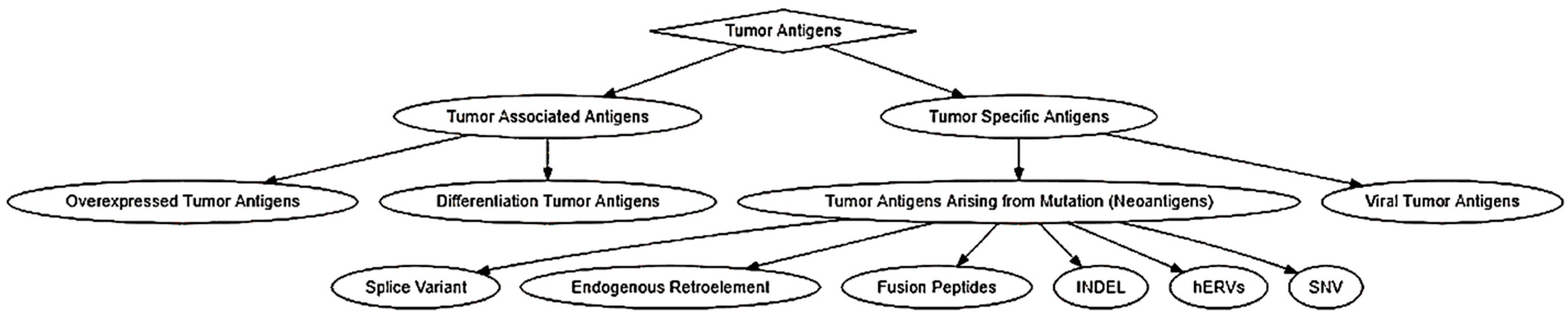
|
Antigen |
MHC-I |
MHC-II |
|---|---|---|
|
α-actinin |
HLA-A2 [54] |
|
|
ARTC1 |
HLA-DR1 [55] |
|
|
B-RAF |
HLA-DR4 [56] |
|
|
β-catenin |
HLA-A24 [57] |
|
|
BCR-ABL |
HLA-A2 [58] HLA-A3 [59] HLA-A11 [60] HLA-B8 [58] |
HLA-DR4 [61] HLA-DR9 [62] HLA-DR11 [63] HLA-DR15 [64] HLA-DR1 [65] |
|
Caspase-5 |
HLA-A2 [66] |
|
|
Caspase-8 |
HLA-B35 [67] |
|
|
CDC27 |
HLA-DR4 [68] |
|
|
CDK-4 |
HLA-A2 [69] |
|
|
CDK-12 |
HLA-A11 [70] |
|
|
CDK2NA |
HLA-A11 [71] |
|
|
CLPP |
HLA-A2 [72] |
|
|
COA-1 |
HLA-DR4 [73] HLA-DR13 [73] |
|
|
CSNK1A1 |
HLA-A2 [70] |
|
|
dek-can |
HLA-DR53 [74] |
|
|
EFTUD2 |
HLA-A3 [75] |
|
|
ELF2M |
HLA-A68 [76] |
|
|
ETV6-AML1 |
HLA-A2 [77] |
|
|
FLT3-ITD |
HLA-A1 [78] |
|
|
fibronectin |
HLA-DR15 [79] |
|
|
FNDC3B |
HLA-A2 [80] |
|
|
GAS7 |
HLA-A2 [70] |
|
|
GPNMB |
HLA-A3 [75] |
|
|
HAUS3 |
HLA-A2 [70] |
|
|
HSDL1 |
HLA-Cw14 [81] |
|
|
HSP70-2 |
HLA-A2 [82] |
|
|
KIA A0205 |
HLA-B44 [83] |
|
|
K-ras |
HLA-B35 [84] HLA-Cw8 [85] |
|
|
LDLR-FUT |
HLA-DR1 [86] |
|
|
MART-2 |
HLA-A1 [87] |
|
|
MATN |
HLA-A11 [70] |
|
|
ME1 |
HLA-A2 [88] |
|
|
MUM-1 |
HLA-B44 [89] |
|
|
MUM-2 |
HLA-B44 [90] HLA-Cw6 [90] |
|
|
MUM-3 |
HLA-A68 [91] |
|
|
Myosin-m |
HLA-A3 [92] |
|
|
N-ras |
HLA-A1 [93] |
|
|
neo-PAP |
HLA-DR7 [94] |
|
|
NFYC |
HLA-B52 [95] |
|
|
OGT |
HLA-A2 [96] |
|
|
OS-9 |
HLA-B44 [97] |
|
|
p14ARF |
HLA-A11 [71] |
|
|
p16INK |
HLA-A11 [71] |
|
|
pml-RARalpha fusion protein |
HLA-DR11 [98] |
|
|
PPP1R3B |
HLA-A1 [70] |
|
|
PRDX5 |
HLA-A2 [99] |
|
|
PTPRK |
HLA-DR10 [100] |
|
|
RBAF600 |
HLA-B7 [75] |
|
|
SIRT2 |
HLA-A3 [75] |
|
|
SNDRP1 |
HLA-B38 |
|
|
SYT-SSX1 or -SSX2 fusion protein |
HLA-B7 [101] |
|
|
TGFBRII |
HLA-DR3 [102] |
|
|
TP53 |
HLA-A2 [103] |
|
|
TPI |
HLA-DR1 [104] |
|
|
Annexin II |
HLA-DR4 [105] |
2.2. Biochemical and Bioinformatic Approaches for the Identification of Tumor Antigens
2.2.1. Serological Analysis of Recombinant Tumor cDNA Libraries (SEREX)
SEREX is a technique developed by Sahin, Pfreundschuh et al., whereby one can identify novel tumor antigens through the sampling of mRNA from fresh tumor specimens, instead of in vitro cancer cell lines [106]. This is because in vitro cancer cell lines can be subject to either a loss or unwanted generation of cancer antigens, due to mutations that might arise during the continuance of the cell culture. The mRNA extracted from the fresh tumor specimen is used to build a cDNA library and subsequently cloned into a λ phage expression vector. This λ phage expression vector is transfected into Escherichia coli for the recombinant expression of potential cancer antigens in the cDNA library. Recombinant proteins are collected and transferred onto a nitrocellulose membrane, blocked, and exposed to autologous diluted serum (1:100 or 1:1000) from the same patient that tumor specimens are taken from. The serum is diluted to ensure that only high-titer IgG antibodies react with the recombinant proteins on the nitrocellulose membrane. Subsequently, a secondary immunoscreening is performed with anti-human IgG for the purification and identification of positive clones while eliminating the false positives that can arise from residual recombinant immunoglobulin (IgG) expression, due to the B-cells and plasma cells that are present in the tumor specimen being sampled. Finally, positive clones are subcloned for isolation of that specific antigenic cDNA fragment and that cDNA fragment is sequenced to determine its nucleotide sequence [107,108,109][107][108][109]. One of the major drawbacks of SEREX is that the bacteria is incapable of expressing low abundant TAAs and their tumor-specific post-translational modification [110,111][110][111]. SEREX-defined antigens are usually weakly immunogenic due to the lack of mutations or structural aberrance [112]. SEREX has also been criticized for its demanding protocol and poor reproducibility [111].2.2.2. Computational Prediction Methods for Cancer Antigens
The traditional pipeline for computational tumor-specific antigen prediction segments itself into five distinct steps: variant calling, HLA typing, peptide enumeration, HLA binding prediction, and finally therapy generation (Figure 2) [30,113][30][113]. Variant calling involves predicting potential cancer antigens through methods that use data from high-throughput genetic sequencing (RNA-Seq or DNA-Seq). This genetic data is processed through algorithms best suited to predict the potential antigenicity of a TSA, depending on its mutational origin: SNV, INDEL, frameshifts, fusion proteins, endogenous retroelement, or hERVs [30,113,114][30][113][114]. HLA typing is performed to determine HLA allele frequencies [30,113,115][30][113][115]. Peptide enumeration is done to determine the peptide sequences of the potential antigenic mutants and to sort them from incompatible antigenic sequences that arise from non-sense mutations and other non-functional genetic aberrations [30,113,116][30][113][116]. HLA binding prediction is enacted to determine the binding of affinity of the antigenic peptide to the corresponding HLA molecule. This HLA-to-peptide affinity quantification is usually expressed as either a ranked percentile or a KD ≤ 500 nM (standard cutoff for detection) [80,117][80][117]. Finally, the genetic information gathered was utilized to make vaccine and cellular therapeutics such as DNA, RNA, peptide, and autologous DC or T-cell vaccines.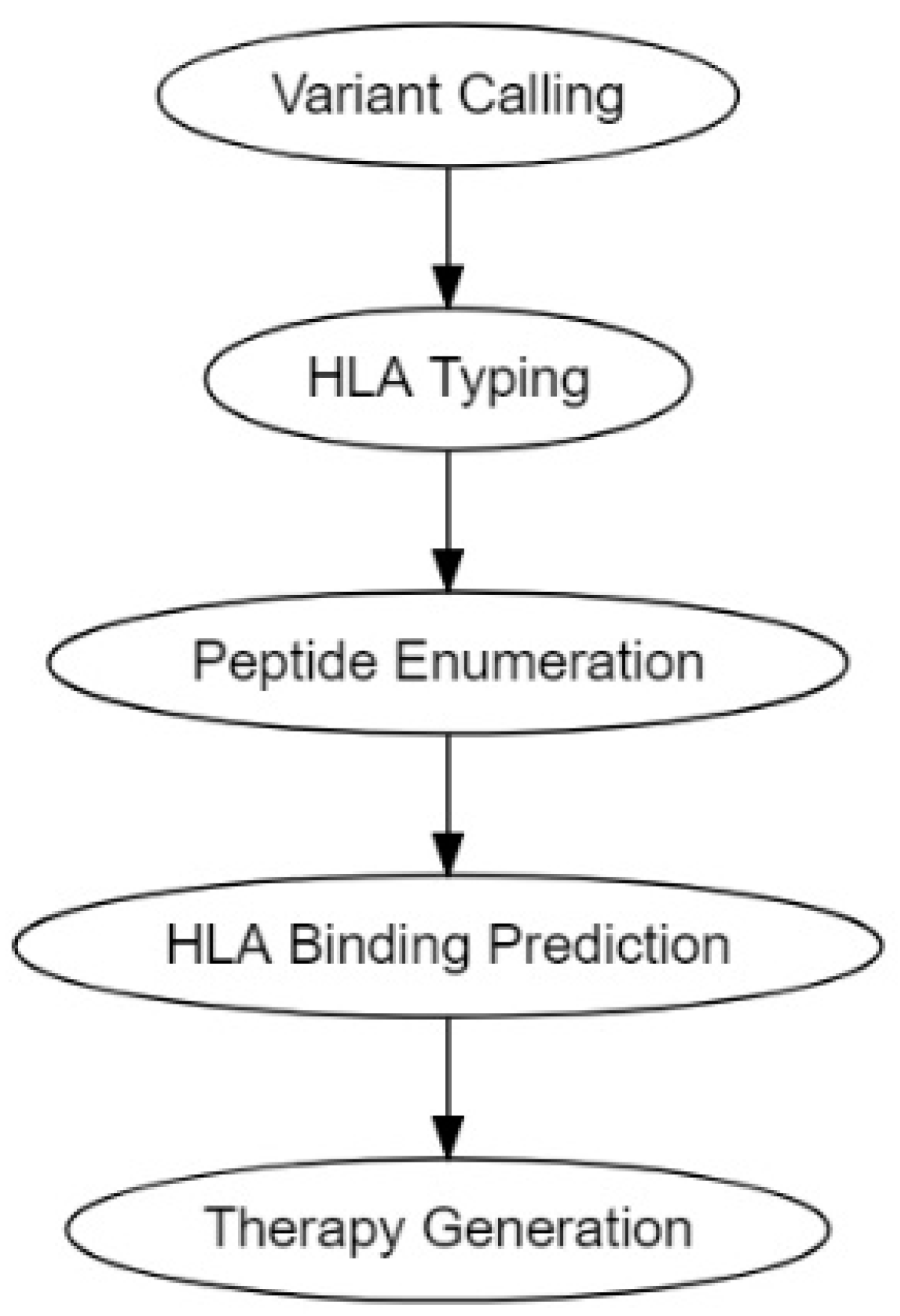
2.3. Delivery of Neoantigens
Neoantigens can be delivered in the form of synthetic long peptides or neoepitope-encoding mRNA or DNA [5]. Direct injection of soluble subunit antigens and immune adjuvants can only induce modest immune responses due to their uncontrolled systemic distribution and poor targeting and accumulation in lymphoid organs. DNA requires electroporation-facilitated delivery and extra processing before presentation by DCs [5,128][5][128]. mRNA needs delivery platforms to facilitate the intracellular delivery and protect it from ribonuclease degradation [129,130][129][130]. Therefore, neoantigen vaccines formulated with novel technologies and biomaterials, such as lipids and biodegradable polymers, are pursued to improve the safety and efficiency of neoantigen delivery. Current progress in the delivery of neoantigen vaccines has been discussed in other reviews [131,132][131][132].3. Vaccine Adjuvants
Adjuvants are known as a variety of substances used in combination with a specific antigen that produce stronger immunity than the antigen alone [133]. Incorporating an adjuvant in a vaccine does not only strengthen the adaptive response to antigens but also enables a comparable response with a lower dose of antigens or less frequent vaccinations relative to unadjuvanted vaccines [134,135][134][135]. As such, adjuvants have become essential components of many successful vaccines and those still in clinical trials. For example, the adjuvant system 04 (AS04), consisting of aluminum salt particles loaded with MPL (3-O-desacyl-4′-monophosphoryl lipid A), has been used in two licensed vaccines, CervarixTM against human papillomavirus (HPV) and FendrixTM against hepatitis B virus [136,137][136][137]. Poly ICLC is a derivative of toll-like receptor (TLR) 3 agonist polyriboinosinic-polyribocytidylic acid (poly(I:C)) stabilized with carboxymethylcellulose and poly-L-lysine [138], which has been widely utilized as an adjuvant in therapeutic vaccines against different cancers in more than 50 clinical trials [139]. From a mechanistic view, an immune response starts with sampling and presentation of antigens by APCs. Evidence has demonstrated that adjuvants can activate APCs, facilitate antigen uptake and cross-presentation between APCs and T-cells, and stimulate the production of immunoregulatory molecules [147,148][140][141]. In addition, the importance of adjuvants is underlined when they are exploited to direct the desired types of immune response (e.g., type-1 immunity versus type-2 immunity, CD8+ versus CD4+ T-cells) and promote the generation of immunological memory [135]. Based on their modes of action, adjuvants can be grouped into two main categories, delivery systems and immune potentiators [141][142]. Delivery systems act as carriers or depots where antigens and other vaccine components can stay and maintain their stability; in the meantime, they create local proinflammatory responses and recruit APCs. Immune potentiators can activate innate immune cells directly or through PRRs, such as TLRs, nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), and retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs) [135]. The interactions between PRRs and pathogen-associated molecular patterns (PAMPs) activate innate immune cells to produce chemokines and cytokines [140][143]. Once activated, APCs will present antigens to T-cells via MHC and release costimulatory molecules to prime naïve T-cells, bridging the fast-acting innate response with antigen-specific adaptive response.References
- Plotkin, S.A. Vaccines: Past, present and future. Nat. Med. 2005, 11, S5–S11.
- Riedel, S. Edward Jenner and the history of smallpox and vaccination. Proc. (Bayl. Univ. Med. Cent.) 2005, 18, 21–25.
- Tregoning, J.S.; Flight, K.E.; Higham, S.L.; Wang, Z.; Pierce, B.F. Progress of the COVID-19 vaccine effort: Viruses, vaccines and variants versus efficacy, effectiveness and escape. Nat. Rev. Immunol. 2021, 21, 626–636.
- A Phase 1 Study to Evaluate the Safety and Immunogenicity of eOD-GT8 60mer mRNA Vaccine (mRNA-1644) and Core-g28v2 60mer mRNA Vaccine (mRNA-1644v2-Core); 2021. Available online: https://clinicaltrials.gov/ct2/show/results/NCT05001373?view=results (accessed on 30 November 2021).
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378.
- Prehn, R.T.; Main, J.M. Immunity to Methylcholanthrene-Induced Sarcomas. J. Natl. Cancer Inst. 1957, 18, 769–778.
- van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991, 254, 1643.
- Murphy, K.; Weaver, C.; Janeway, C. Janeway’s Immunobiology, 9th ed.; Garland Science: New York, NY, USA, 2017.
- Piertney, S.B.; Oliver, M.K. The evolutionary ecology of the major histocompatibility complex. Heredity 2006, 96, 7–21.
- Klein, J.; Figueroa, F. Evolution of the major histocompatibility complex. Crit. Rev. Immunol. 1986, 6, 295–386.
- Altuvia, Y.; Margalit, H. A structure-based approach for prediction of MHC-binding peptides. Methods 2004, 34, 454–459.
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292.
- Jeffery, K.J.M.; Bangham, C.R.M. Do infectious diseases drive MHC diversity? Microbes Infect. 2000, 2, 1335–1341.
- Dausset, J. The Major Histocompatibility Complex in Man. Science 1981, 213, 1469–1474.
- Bruggen, P.V.D.; Szikora, J.-P.; Boël, P.; Wildmann, C.; Somville, M.; Sensi, M.; Boon, T. Autologous cytolytic T lymphocytes recognize a MAGE-1 nonapeptide on melanomas expressing HLA-Cw* 1601. Eur. J. Immunol. 1994, 24, 2134–2140.
- Robbins, P.F.; Kawakami, Y. Human tumor antigens recognized by T cells. Curr. Opin. Immunol. 1996, 8, 628–636.
- Boon, T.; Cerottini, J.-C.; Van den Eynde, B.; van der Bruggen, P.; Van Pel, A. Tumor Antigens Recognized by T Lymphocytes. Annu. Rev. Immunol. 1994, 12, 337–365.
- Bennett, S.R.M.; Carbone, F.R.; Karamalis, F.; Flavell, R.A.; Miller, J.F.A.P.; Heath, W.R. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 1998, 393, 478–480.
- Ridge, J.P.; Di Rosa, F.; Matzinger, P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature 1998, 393, 474–478.
- Schoenberger, S.P.; Toes, R.E.M.; van der Voort, E.I.H.; Offringa, R.; Melief, C.J.M. T-cell help for cytotoxic T lymphocytes is mediated by CD40–CD40L interactions. Nature 1998, 393, 480–483.
- Kennedy, R.; Celis, E. Multiple roles for CD4+ T cells in anti-tumor immune responses. Immunol. Rev. 2008, 222, 129–144.
- Pardoll, D.M.; Topalian, S.L. The role of CD4+ T cell responses in antitumor immunity. Curr. Opin. Immunol. 1998, 10, 588–594.
- Valilou, S.F.; Rezaei, N. Chapter 4—Tumor Antigens. In Vaccines for Cancer Immunotherapy; Rezaei, N., Keshavarz-Fathi, M., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 61–74.
- Boon, T.; Coulie, P.G.; Van den Eynde, B. Tumor antigens recognized by T cells. Immunol. Today 1997, 18, 267–268.
- Nagorsen, D.; Marincola, F.M. Analyzing T Cell Responses: How to Analyze Cellular Immune Responses against Tumor Associated Antigens; Springer: Berlin/Heidelberg, Germany, 2006.
- Wang, R.-F.; Rosenberg, S.A. Human tumor antigens for cancer vaccine development. Immunol. Rev. 1999, 170, 85–100.
- Richards, J.M.; Mehta, N.; Ramming, K.; Skosey, P. Sequential Chemoimmunotherapy in the Treatment of Metastatic Melanoma. In Proceedings of the Cytokines in Hemopoiesis, Oncology, and AIDS II, Berlin/Heidelberg, Germany, 1 August 1992; pp. 721–727.
- Agyemang, A.F.; Odunsi, K.O. Chapter 5—The use of immunotherapy for treatment of chemoresistant ovarian cancer. In Overcoming Ovarian Cancer Chemoresistance; Samimi, G., Annunziata, C., Eds.; Academic Press: Cambridge, MA, USA, 2021; Volume 11, pp. 79–96.
- Gjerstorff, M.F.; Andersen, M.H.; Ditzel, H.J. Oncogenic cancer/testis antigens: Prime candidates for immunotherapy. Oncotarget 2015, 6, 15772–15787.
- Smith, C.C.; Selitsky, S.R.; Chai, S.; Armistead, P.M.; Vincent, B.G.; Serody, J.S. Alternative tumour-specific antigens. Nat. Rev. Cancer 2019, 19, 465–478.
- Goodier, J.L.; Kazazian, H.H. Retrotransposons Revisited: The Restraint and Rehabilitation of Parasites. Cell 2008, 135, 23–35.
- Cherkasova, E.; Scrivani, C.; Doh, S.; Weisman, Q.; Takahashi, Y.; Harashima, N.; Yokoyama, H.; Srinivasan, R.; Linehan, W.M.; Lerman, M.I.; et al. Detection of an Immunogenic HERV-E Envelope with Selective Expression in Clear Cell Kidney Cancer. Cancer Res. 2016, 76, 2177–2185.
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021.
- Jayasinghe, R.G.; Cao, S.; Gao, Q.; Wendl, M.C.; Vo, N.S.; Reynolds, S.M.; Zhao, Y.; Climente-González, H.; Chai, S.; Wang, F.; et al. Systematic Analysis of Splice-Site-Creating Mutations in Cancer. Cell Rep. 2018, 23, 270–281.e273.
- Rowley, J.D. A New Consistent Chromosomal Abnormality in Chronic Myelogenous Leukaemia identified by Quinacrine Fluorescence and Giemsa Staining. Nature 1973, 243, 290–293.
- Nowell, P. The minute chromosome (Phl) in chronic granulocytic leukemia. Blut 1962, 8, 65–66.
- Shepherd, P.; Suffolk, R.; Halsey, J.; Allan, N. Analysis of molecular breakpoint and m-RNA transcripts in a prospective randomized trial of interferon in chronic myeloid leukaemia: No correlation with clinical features, cytogenetic response, duration of chronic phase, or survival. Br. J. Haematol. 1995, 89, 546–554.
- Westbrook, C.A.; Hooberman, A.L.; Spino, C.; Dodge, R.K.; Larson, R.A.; Davey, F.; Wurster-Hill, D.H.; Sobol, R.E.; Schiffer, C.; Bloomfield, C.D. Clinical Significance of the BCR-ABL Fusion Gene in Adult Acute Lymphoblastic Leukemia: A Cancer and Leukemia Group B Study (8762). Blood 1992, 80, 2983–2990.
- Russo, C.; Carroll, A.; Kohler, S.; Borowitz, M.; Amylon, M.; Homans, A.; Kedar, A.; Shuster, J.; Land, V.; Crist, W.; et al. Philadelphia Chromosome and Monosomy 7 in Childhood Acute Lymphoblastic Leukemia: A Pediatric Oncology Group Study. Blood 1991, 77, 1050–1056.
- Suryanarayan, K.; Hunger, S.P.; Kohler, S.; Carroll, A.J.; Crist, W.; Link, M.P.; Cleary, M.L. Consistent Involvement of the BCR Gene by 9;22 Breakpoints in Pediatric Acute Leukemias. Blood 1991, 77, 324–330.
- Kurzrock, R.; Shtalrid, M.; Talpaz, M.; Kloetzer, W.S.; Gutterman, J.U. Expression of c-abl in Philadelphia-Positive Acute Myelogenous Leukemia. Blood 1987, 70, 1584–1588.
- Secker-Walker, L.M.; Morgan, G.J.; Min, T.; John Swansbury, G.; Craig, J.; Yamada, T.; Desalvo, L.; Medina, J.W.; Chowdhury, V.; Donahue, R.P.; et al. Inversion of chromosome 16 with the Philadelphia chromosome in acute myelomonocytic leukemia with eosinophilia: Report of two cases. Cancer Genet. Cytogenet. 1992, 58, 29–34.
- Alimena, G.; Cedrone, M.; Nanni, M.; De Cuia, M.; De Sanctis, V.; Cimino, G.; Mancini, M. Acute leukemia presenting a variant Ph chromosome with p190 expression, dup 3q and-7, developed after malignant lymphoma treated with alkylating agents and topoisomerase II inhibitors. Leukemia 1995, 9, 1483–1486.
- Fujii, H.; Yashige, H.; Misawa, S.; Tanaka, S.; Urata, Y.; Matuyama, F. Ph chromosome in a patient with non-leukemic non-hodgkin B-cell lymphoma. Am. J. Hematol. 1990, 35, 213–215.
- Mitani, K.; Sato, Y.; Tojo, A.; Ishikawa, F.; Kobayashi, Y.; Miura, Y.; Miyazono, K.; And, A.U.; Takaku, F. Philadelphia chromosome positive B-cell type malignant lymphoma expressing an aberrant 190 kDa bcr-abl protein. Br. J. Haematol. 1990, 76, 221–225.
- Juliusson, G.; Friberg, K.; Gahrton, G. Ph chromosome and B-cell malignancy-associated chromosomal aberrations in non-leukaemic immunoblastic B-cell lymphoma. Acta Haematol. 1985, 74, 171–174.
- Martiat, P.; Mecucci, C.; Nizet, Y.; Stul, M.; Philippe, M.; Cassiman, J.-J.; Michaux, J.L.; Van den Berghe, H.; Sokal, G. P190 BCR/ABL transcript in a case of Philadelphia-positive multiple myeloma. Leukemia 1990, 4, 751–754.
- Van Den Berghe, H.a.; Louwagie, A.; Broeckaert-Van Orshoven, A.; David, G.; Verwilghen, R.; Michaux, J.; Sokal, G. Philadelphia chromosome in human multiple myeloma. J. Natl. Cancer Inst. 1979, 63, 11–16.
- Karpas, A.; Fischer, P.; Swirsky, D. Human myeloma cell line carrying a Philadelphia chromosome. Science 1982, 216, 997–999.
- Bos, J.L. Ras Oncogenes in Human Cancer: A Review. Cancer Res. 1989, 49, 4682–4689.
- Bourgault Villada, I.; Bénéton, N.; Bony, C.; Connan, F.; Monsonego, J.; Bianchi, A.; Saiag, P.; Lévy, J.P.; Guillet, J.G.; Choppin, J. Identification in humans of HPV-16 E6 and E7 protein epitopes recognized by cytolytic T lymphocytes in association with HLA-B18 and determination of the HLA-B18-specific binding motif. Eur. J. Immunol. 2000, 30, 2281–2289.
- Peng, S.; Trimble, C.; Wu, L.; Pardoll, D.; Roden, R.; Hung, C.-F.; Wu, T.C. HLA-DQB1*02–Restricted HPV-16 E7 Peptide–Specific CD4+ T-Cell Immune Responses Correlate with Regression of HPV-16–Associated High-Grade Squamous Intraepithelial Lesions. Clin. Cancer Res. 2007, 13, 2479.
- Kaufmann, A.M.; Nieland, J.; Schinz, M.; Nonn, M.; Gabelsberger, J.; Meissner, H.; Müller, R.T.; Jochmus, I.; Gissmann, L.; Schneider, A.; et al. HPV16 L1E7 chimeric virus-like particles induce specific HLA-restricted T cells in humans after in vitro vaccination. Int. J. Cancer 2001, 92, 285–293.
- Echchakir, H.; Mami-Chouaib, F.; Vergnon, I.; Baurain, J.-F.; Karanikas, V.; Chouaib, S.; Coulie, P.G. A Point Mutation in the α-actinin-4 Gene Generates an Antigenic Peptide Recognized by Autologous Cytolytic T Lymphocytes on a Human Lung Carcinoma. Cancer Res. 2001, 61, 4078.
- Wang, H.Y.; Peng, G.; Guo, Z.; Shevach, E.M.; Wang, R.-F. Recognition of a New ARTC1 Peptide Ligand Uniquely Expressed in Tumor Cells by Antigen-Specific CD4+ Regulatory T Cells. J. Immunol. 2005, 174, 2661.
- Sharkey, M.S.; Lizée, G.; Gonzales, M.I.; Patel, S.; Topalian, S.L. CD4+ T-Cell Recognition of Mutated B-RAF in Melanoma Patients Harboring the V599E Mutation. Cancer Res. 2004, 64, 1595.
- Robbins, P.F.; El-Gamil, M.; Li, Y.F.; Kawakami, Y.; Loftus, D.; Appella, E.; Rosenberg, S.A. A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J. Exp. Med. 1996, 183, 1185–1192.
- Yotnda, P.; Firat, H.; Garcia-Pons, F.; Garcia, Z.; Gourru, G.; Vernant, J.P.; Lemonnier, F.A.; Leblond, V.; Langlade-Demoyen, P. Cytotoxic T cell response against the chimeric p210 BCR-ABL protein in patients with chronic myelogenous leukemia. J. Clin. Investig. 1998, 101, 2290–2296.
- Greco, G.; Fruci, D.; Accapezzato, D.; Barnaba, V.; Nisini, R.; Alimena, G.; Montefusco, E.; Vigneti, E.; Butler, R.; Tanigaki, N.; et al. Two bcr-abl junction peptides bind HLA-A3 molecules and allow specific induction of human cytotoxic T lymphocytes. Leukemia 1996, 10, 693–699.
- Bocchia, M.; Korontsvit, T.; Xu, Q.; Mackinnon, S.; Yang, S.; Sette, A.; Scheinberg, D. Specific human cellular immunity to bcr-abl oncogene-derived peptides. Blood 1996, 87, 3587–3592.
- Bosch, G.; Joosten, A.; Kessler, J.; Melief, C.; Leeksma, O. Recognition of BCR-ABL positive leukemic blasts by human CD4+ T cells elicited by primary in vitro immunization with a BCR-ABL breakpoint peptide. Blood 1996, 88, 3522–3527.
- Yasukawa, M.; Ohminami, H.; Kaneko, S.; Yakushijin, Y.; Nishimura, Y.; Inokuchi, K.; Miyakuni, T.; Nakao, S.; Kishi, K.; Kubonishi, I.; et al. CD4+ Cytotoxic T-Cell Clones Specific for bcr-abl b3a2 Fusion Peptide Augment Colony Formation by Chronic Myelogenous Leukemia Cells in a b3a2-Specific and HLA-DR–Restricted Manner. Blood 1998, 92, 3355–3361.
- Pawelec, G.; Max, H.; Halder, T.; Bruserud, O.; Merl, A.; da Silva, P.; Kalbacher, H. BCR/ABL leukemia oncogene fusion peptides selectively bind to certain HLA-DR alleles and can be recognized by T cells found at low frequency in the repertoire of normal donors. Blood 1996, 88, 2118–2124.
- Tanaka, Y.; Takahashi, T.; Nieda, M.; Masuda, S.; Kashiwase, K.; Ogawa, S.; Chiba, S.; Juji, T.; Hirai, H. Generation of HLA-DRB1*1501-restricted p190 minor bcr–abl (e1a2)-specific CD4+ T lymphocytes. Br. J. Haematol. 2000, 109, 435–437.
- ten Bosch, G.J.A.; Kessler, J.H.; Joosten, A.M.; Bres-Vloemans, A.A.; Geluk, A.; Godthelp, B.C.; van Bergen, J.; Melief, C.J.M.; Leeksma, O.C. A BCR-ABL Oncoprotein p210b2a2 Fusion Region Sequence Is Recognized by HLA-DR2a Restricted Cytotoxic T Lymphocytes and Presented by HLA-DR Matched Cells Transfected With an Iib2a2 Construct. Blood 1999, 94, 1038–1045.
- Schwitalle, Y.; Linnebacher, M.; Ripberger, E.; Gebert, J.; von Knebel Doeberitz, M. Immunogenic peptides generated by frameshift mutations in DNA mismatch repair-deficient cancer cells. Cancer Immun. Arch. 2004, 4, 14.
- Mandruzzato, S.; Brasseur, F.; Andry, G.; Boon, T.; van der Bruggen, P. A CASP-8 mutation recognized by cytolytic T lymphocytes on a human head and neck carcinoma. J. Exp. Med. 1997, 186, 785–793.
- Wang, R.-F.; Wang, X.; Atwood, A.C.; Topalian, S.L.; Rosenberg, S.A. Cloning Genes Encoding MHC Class II-Restricted Antigens: Mutated CDC27 as a Tumor Antigen. Science 1999, 284, 1351–1354.
- Wolfel, T.; Hauer, M.; Schneider, J.; Serrano, M.; Wolfel, C.; Klehmann-Hieb, E.; De Plaen, E.; Hankeln, T.; Meyer zum Buschenfelde, K.; Beach, D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 1995, 269, 1281–1284.
- Robbins, P.F.; Lu, Y.-C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752.
- Huang, J.; El-Gamil, M.; Dudley, M.E.; Li, Y.F.; Rosenberg, S.A.; Robbins, P.F. T cells associated with tumor regression recognize frameshifted products of the CDKN2A tumor suppressor gene locus and a mutated HLA class I gene product. J. Immunol. 2004, 172, 6057–6064.
- Corbière, V.; Chapiro, J.; Stroobant, V.; Ma, W.; Lurquin, C.; Lethé, B.; van Baren, N.; Van den Eynde, B.J.; Boon, T.; Coulie, P.G. Antigen Spreading Contributes to MAGE Vaccination-Induced Regression of Melanoma Metastases. Cancer Res. 2011, 71, 1253.
- Maccalli, C.; Pende, D.; Castelli, C.; Mingari, M.C.; Robbins, P.F.; Parmiani, G. NKG2D engagement of colorectal cancer-specific T cells strengthens TCR-mediated antigen stimulation and elicits TCR independent anti-tumor activity. Eur. J. Immunol. 2003, 33, 2033–2043.
- Makita, M.; Azuma, T.; Hamaguchi, H.; Niiya, H.; Kojima, K.; Fujita, S.; Tanimoto, M.; Harada, M.; Yasukawa, M. Leukemia-associated fusion proteins, dek-can and bcr-abl, represent immunogenic HLA-DR-restricted epitopes recognized by fusion peptide-specific CD4+ T lymphocytes. Leukemia 2002, 16, 2400–2407.
- Lennerz, V.; Fatho, M.; Gentilini, C.; Frye, R.A.; Lifke, A.; Ferel, D.; Wölfel, C.; Huber, C.; Wölfel, T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl. Acad. Sci. USA 2005, 102, 16013–16018.
- Hogan, K.T.; Eisinger, D.P.; Cupp, S.B.; Lekstrom, K.J.; Deacon, D.D.; Shabanowitz, J.; Hunt, D.F.; Engelhard, V.H.; Slingluff, C.L.; Ross, M.M. The Peptide Recognized by HLA-A68.2-restricted, Squamous Cell Carcinoma of the Lung-specific Cytotoxic T Lymphocytes Is Derived from a Mutated Elongation Factor 2 Gene. Cancer Res. 1998, 58, 5144.
- Yotnda, P.; Garcia, F.; Peuchmaur, M.; Grandchamp, B.; Duval, M.; Lemonnier, F.; Vilmer, E.; Langlade-Demoyen, P. Cytotoxic T cell response against the chimeric ETV6-AML1 protein in childhood acute lymphoblastic leukemia. J. Clin. Investig. 1998, 102, 455–462.
- Graf, C.; Heidel, F.; Tenzer, S.; Radsak, M.P.; Solem, F.K.; Britten, C.M.; Huber, C.; Fischer, T.; Wölfel, T. A neoepitope generated by an FLT3 internal tandem duplication (FLT3-ITD) is recognized by leukemia-reactive autologous CD8+ T cells. Blood 2006, 109, 2985–2988.
- Wang, H.Y.; Zhou, J.; Zhu, K.; Riker, A.I.; Marincola, F.M.; Wang, R.-F. Identification of a mutated fibronectin as a tumor antigen recognized by CD4+ T cells: Its role in extracellular matrix formation and tumor metastasis. J. Exp. Med. 2002, 195, 1397–1406.
- Rajasagi, M.; Shukla, S.A.; Fritsch, E.F.; Keskin, D.B.; DeLuca, D.; Carmona, E.; Zhang, W.; Sougnez, C.; Cibulskis, K.; Sidney, J.; et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood 2014, 124, 453–462.
- Wick, D.A.; Webb, J.R.; Nielsen, J.S.; Martin, S.D.; Kroeger, D.R.; Milne, K.; Castellarin, M.; Twumasi-Boateng, K.; Watson, P.H.; Holt, R.A.; et al. Surveillance of the Tumor Mutanome by T Cells during Progression from Primary to Recurrent Ovarian Cancer. Clin. Cancer Res. 2014, 20, 1125.
- Gaudin, C.; Kremer, F.; Angevin, E.; Scott, V.; Triebel, F. A hsp70-2 Mutation Recognized by CTL on a Human Renal Cell Carcinoma. J. Immunol. 1999, 162, 1730.
- Guéguen, M.; Patard, J.-J.; Gaugler, B.; Brasseur, F.; Renauld, J.-C.; Van Cangh, P.J.; Boon, T.; Van den Eynde, B.t.J. An Antigen Recognized by Autologous CTLs on a Human Bladder Carcinoma. J. Immunol. 1998, 160, 6188.
- Gjertsen, M.K.; Bjørheim, J.; Saeterdal, I.; Myklebust, J.; Gaudernack, G. Cytotoxic CD4+ and CD8+ T lymphocytes, generated by mutant p21-ras (12VAL) peptide vaccination of a patient, recognize 12VAL-dependent nested epitopes present within the vaccine peptide and kill autologous tumour cells carrying this mutation. Int. J. Cancer 1997, 72, 784–790.
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262.
- Wang, R.F.; Wang, X.; Rosenberg, S.A. Identification of a novel major histocompatibility complex class II-restricted tumor antigen resulting from a chromosomal rearrangement recognized by CD4(+) T cells. J. Exp. Med. 1999, 189, 1659–1668.
- Kawakami, Y.; Wang, X.; Shofuda, T.; Sumimoto, H.; Tupesis, J.P.; Fitzgerald, E.; Rosenberg, S.A. Isolation of a New Melanoma Antigen, MART-2, Containing a Mutated Epitope Recognized by Autologous Tumor-Infiltrating T Lymphocytes. J. Immunol. 2001, 166, 2871.
- Karanikas, V.; Colau, D.; Baurain, J.-F.; Chiari, R.; Thonnard, J.; Gutierrez-Roelens, I.; Goffinet, C.; Schaftingen, E.V.; Weynants, P.; Boon, T.; et al. High Frequency of Cytolytic T Lymphocytes Directed against a Tumor-specific Mutated Antigen Detectable with HLA Tetramers in the Blood of a Lung Carcinoma Patient with Long Survival. Cancer Res. 2001, 61, 3718.
- Coulie, P.G.; Lehmann, F.; Lethé, B.; Herman, J.; Lurquin, C.; Andrawiss, M.; Boon, T. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc. Natl. Acad. Sci. USA 1995, 92, 7976–7980.
- Chiari, R.; Foury, F.; De Plaen, E.; Baurain, J.-F.; Thonnard, J.; Coulie, P.G. Two Antigens Recognized by Autologous Cytolytic T Lymphocytes on a Melanoma Result from a Single Point Mutation in an Essential Housekeeping Gene. Cancer Res. 1999, 59, 5785.
- Baurain, J.-F.; Colau, D.; van Baren, N.; Landry, C.; Martelange, V.; Vikkula, M.; Boon, T.; Coulie, P.G. High Frequency of Autologous Anti-Melanoma CTL Directed Against an Antigen Generated by a Point Mutation in a New Helicase Gene. J. Immunol. 2000, 164, 6057.
- Zorn, E.; Hercend, T. A natural cytotoxic T cell response in a spontaneously regressing human melanoma targets a neoantigen resulting from a somatic point mutation. Eur. J. Immunol. 1999, 29, 592–601.
- Linard, B.; Bézieau, S.; Benlalam, H.; Labarrière, N.; Guilloux, Y.; Diez, E.; Jotereau, F. A ras-Mutated Peptide Targeted by CTL Infiltrating a Human Melanoma Lesion. J. Immunol. 2002, 168, 4802.
- Topalian, S.L.; Gonzales, M.I.; Ward, Y.; Wang, X.; Wang, R.-F. Revelation of a Cryptic Major Histocompatibility Complex Class II-restricted Tumor Epitope in a Novel RNA-processing Enzyme. Cancer Res. 2002, 62, 5505.
- Takenoyama, M.; Baurain, J.-F.; Yasuda, M.; So, T.; Sugaya, M.; Hanagiri, T.; Sugio, K.; Yasumoto, K.; Boon, T.; Coulie, P.G. A point mutation in the NFYC gene generates an antigenic peptide recognized by autologous cytolytic T lymphocytes on a human squamous cell lung carcinoma. Int. J. Cancer 2006, 118, 1992–1997.
- Ripberger, E.; Linnebacher, M.; Schwitalle, Y.; Gebert, J.; Doeberitz, M.V.K. Identification of an HLA-A0201-Restricted CTL Epitope Generated by a Tumor-Specific Frameshift Mutation in a Coding Microsatellite of the OGT Gene. J. Clin. Immunol. 2003, 23, 415–423.
- Vigneron, N.; Ooms, A.; Morel, S.; Degiovanni, G.; Van Den Eynde, B.J. Identification of a new peptide recognized by autologous cytolytic T lymphocytes on a human melanoma. Cancer Immun. 2002, 2, 9.
- Gambacorti-Passerini, C.; Grignani, F.; Arienti, F.; Pandolfi, P.P.; Pelicci, P.G.; Parmiani, G. Human CD4 lymphocytes specifically recognize a peptide representing the fusion region of the hybrid protein pml/RAR alpha present in acute promyelocytic leukemia cells. Blood 1993, 81, 1369–1375.
- Sensi, M.; Nicolini, G.; Zanon, M.; Colombo, C.; Molla, A.; Bersani, I.; Lupetti, R.; Parmiani, G.; Anichini, A. Immunogenicity without Immunoselection: A Mutant but Functional Antioxidant Enzyme Retained in a Human Metastatic Melanoma and Targeted by CD8+ T Cells with a Memory Phenotype. Cancer Res. 2005, 65, 632.
- Novellino, L.; Renkvist, N.; Rini, F.; Mazzocchi, A.; Rivoltini, L.; Greco, A.; Deho, P.; Squarcina, P.; Robbins, P.F.; Parmiani, G.; et al. Identification of a Mutated Receptor-Like Protein Tyrosine Phosphatase κ as a Novel, Class II HLA-Restricted Melanoma Antigen. J. Immunol. 2003, 170, 6363–6370.
- Worley, B.S.; van den Broeke, L.T.; Goletz, T.J.; Pendleton, C.D.; Daschbach, E.M.; Thomas, E.K.; Marincola, F.M.; Helman, L.J.; Berzofsky, J.A. Antigenicity of Fusion Proteins from Sarcoma-associated Chromosomal Translocations. Cancer Res. 2001, 61, 6868.
- Sæterdal, I.; Bjørheim, J.; Lislerud, K.; Gjertsen, M.K.; Bukholm, I.K.; Olsen, O.C.; Nesland, J.M.; Eriksen, J.A.; Møller, M.; Lindblom, A.; et al. Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 13255–13260.
- Malekzadeh, P.; Pasetto, A.; Robbins, P.F.; Parkhurst, M.R.; Paria, B.C.; Jia, L.; Gartner, J.J.; Hill, V.; Yu, Z.; Restifo, N.P.; et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Investig. 2019, 129, 1109–1114.
- Pieper, R.; Christian, R.E.; Gonzales, M.I.; Nishimura, M.I.; Gupta, G.; Settlage, R.E.; Shabanowitz, J.; Rosenberg, S.A.; Hunt, D.F.; Topalian, S.L. Biochemical identification of a mutated human melanoma antigen recognized by CD4(+) T cells. J. Exp. Med. 1999, 189, 757–766.
- Li, K.; Adibzadeh, M.; Halder, T.; Kalbacher, H.; Heinzel, S.; Müller, C.; Zeuthen, J.; Pawelec, G. Tumour-specific MHC-class-II-restricted responses after in vitro sensitization to synthetic peptides corresponding to gp100 and Annexin II eluted from melanoma cells. Cancer Immunol. Immunother. 1998, 47, 32–38.
- Sahin, U.; Türeci, O.; Schmitt, H.; Cochlovius, B.; Johannes, T.; Schmits, R.; Stenner, F.; Luo, G.; Schobert, I.; Pfreundschuh, M. Human neoplasms elicit multiple specific immune responses in the autologous host. Proc. Natl. Acad. Sci. USA 1995, 92, 11810–11813.
- Sahin, U.; Pfreundschuh, M. Serological analysis of human tumor antigens: Molecular definition and implications. Mol. Med. Today 1997, 3, 342–349.
- Chen, Y.-T.; Gure, A.O.; Scanlan, M.J. Serological Analysis of Expression cDNA Libraries (SEREX). In Pancreatic Cancer; Springer: Berlin/Heidelberg, Germany, 2005; pp. 207–216.
- Türeci, Ö.; Usener, D.; Schneider, S.; Sahin, U. Identification of tumor-associated autoantigens with SEREX. In Adoptive Immunotherapy: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2005; pp. 137–154.
- Rauch, J.; Gires, O. SEREX, Proteomex, AMIDA, and beyond: Serological screening technologies for target identification. PROTEOMICS—Clin. Appl. 2008, 2, 355–371.
- Whittemore, K.; Sykes, K. A microarray method for identifying tumor antigens by screening a tumor cDNA expression library against cancer sera. Hum. Vaccines Immunother. 2013, 9, 2178–2188.
- Boncheva, V.; Bonney, S.A.; Brooks, S.E.; Tangney, M.; O’Sullivan, G.; Mirnezami, A.; Guinn, B.A. New targets for the immunotherapy of colon cancer—does reactive disease hold the answer? Cancer Gene Ther. 2013, 20, 157–168.
- Roudko, V.; Greenbaum, B.; Bhardwaj, N. Computational Prediction and Validation of Tumor-Associated Neoantigens. Front. Immunol. 2020, 11, 27.
- Koboldt, D.C. Best practices for variant calling in clinical sequencing. Genome Med. 2020, 12, 91.
- Orenbuch, R.; Filip, I.; Rabadan, R. HLA Typing from RNA Sequencing and Applications to Cancer. In Bioinformatics for Cancer Immunotherapy: Methods and Protocols; Boegel, S., Ed.; Springer: New York, NY, USA, 2020; pp. 71–92.
- Díaz-Eufracio, B.I.; Palomino-Hernández, O.; Arredondo-Sánchez, A.; Medina-Franco, J.L. D-Peptide Builder: A Web Service to Enumerate, Analyze, and Visualize the Chemical Space of Combinatorial Peptide Libraries. Mol. Inform. 2020, 39, 2000035.
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221.
- Bjerregaard, A.-M.; Nielsen, M.; Hadrup, S.R.; Szallasi, Z.; Eklund, A.C. MuPeXI: Prediction of neo-epitopes from tumor sequencing data. Cancer Immunol. Immunother. 2017, 66, 1123–1130.
- Chang, T.-C.; Carter, R.A.; Li, Y.; Li, Y.; Wang, H.; Edmonson, M.N.; Chen, X.; Arnold, P.; Geiger, T.L.; Wu, G.; et al. The neoepitope landscape in pediatric cancers. Genome Med. 2017, 9, 78.
- Hundal, J.; Carreno, B.M.; Petti, A.A.; Linette, G.P.; Griffith, O.L.; Mardis, E.R.; Griffith, M. pVAC-Seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Med. 2016, 8, 11.
- Kim, S.; Kim, H.S.; Kim, E.; Lee, M.G.; Shin, E.C.; Paik, S.; Kim, S. Neopepsee: Accurate genome-level prediction of neoantigens by harnessing sequence and amino acid immunogenicity information. Ann. Oncol. 2018, 29, 1030–1036.
- Rech, A.J.; Balli, D.; Mantero, A.; Ishwaran, H.; Nathanson, K.L.; Stanger, B.Z.; Vonderheide, R.H. Tumor Immunity and Survival as a Function of Alternative Neopeptides in Human Cancer. Cancer Immunol. Res. 2018, 6, 276–287.
- Rubinsteyn, A.; Kodysh, J.; Hodes, I.; Mondet, S.; Aksoy, B.A.; Finnigan, J.P.; Bhardwaj, N.; Hammerbacher, J. Computational Pipeline for the PGV-001 Neoantigen Vaccine Trial. Front. Immunol. 2018, 8, 1807.
- Zhang, J.; Mardis, E.R.; Maher, C.A. INTEGRATE-neo: A pipeline for personalized gene fusion neoantigen discovery. Bioinformatics 2017, 33, 555–557.
- Zhou, Z.; Lyu, X.; Wu, J.; Yang, X.; Wu, S.; Zhou, J.; Gu, X.; Su, Z.; Chen, S. TSNAD: An integrated software for cancer somatic mutation and tumour-specific neoantigen detection. R. Soc. Open Sci. 2017, 4, 170050.
- Smit, A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. Available online: http://www.repeatmasker.org (accessed on 30 November 2021).
- Smith, C.C.; Beckermann, K.E.; Bortone, D.S.; De Cubas, A.A.; Bixby, L.M.; Lee, S.J.; Panda, A.; Ganesan, S.; Bhanot, G.; Wallen, E.M.; et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J. Clin. Investig. 2018, 128, 4804–4820.
- Lee, S.-H.; Danishmalik, S.N.; Sin, J.-I. DNA vaccines, electroporation and their applications in cancer treatment. Hum. Vaccines Immunother. 2015, 11, 1889–1900.
- Zhao, Z.; Zheng, L.; Chen, W.; Weng, W.; Song, J.; Ji, J. Delivery strategies of cancer immunotherapy: Recent advances and future perspectives. J. Hematol. Oncol. 2019, 12, 126.
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279.
- Guo, Y.; Lei, K.; Tang, L. Neoantigen Vaccine Delivery for Personalized Anticancer Immunotherapy. Front. Immunol. 2018, 9, 1499.
- Um, W.; Gupta, A.; Song, S.H.; Kim, C.H.; Park, J.H. Biomaterials as Antigen Delivery Carrier for Cancer Immunotherapy. Macromol. Res. 2021, 29, 834–842.
- Cox, J.C.; Coulter, A.R. Adjuvants—A classification and review of their modes of action. Vaccine 1997, 15, 248–256.
- Banzhoff, A.; Gasparini, R.; Laghi-Pasini, F.; Staniscia, T.; Durando, P.; Montomoli, E.; Capecchi, P.L.; di Giovanni, P.; Sticchi, L.; Gentile, C.; et al. MF59-adjuvanted H5N1 vaccine induces immunologic memory and heterotypic antibody responses in non-elderly and elderly adults. PLoS ONE 2009, 4, e4384.
- Coffman, R.L.; Sher, A.; Seder, R.A. Vaccine Adjuvants: Putting Innate Immunity to Work. Immunity 2010, 33, 492–503.
- Didierlaurent, A.M.; Morel, S.; Lockman, L.; Giannini, S.L.; Bisteau, M.; Carlsen, H.; Kielland, A.; Vosters, O.; Vanderheyde, N.; Schiavetti, F.; et al. AS04, an Aluminum Salt- and TLR4 Agonist-Based Adjuvant System, Induces a Transient Localized Innate Immune Response Leading to Enhanced Adaptive Immunity. J. Immunol. 2009, 183, 6186–6197.
- Garçon, N.; Di Pasquale, A. From discovery to licensure, the Adjuvant System story. Hum. Vaccines Immunother. 2016, 13, 19–33.
- Ammi, R.; De Waele, J.; Willemen, Y.; Van Brussel, I.; Schrijvers, D.M.; Lion, E.; Smits, E.L.J. Poly(I:C) as cancer vaccine adjuvant: Knocking on the door of medical breakthroughs. Pharmacol. Ther. 2015, 146, 120–131.
- Apostólico, J.d.S.; Lunardelli, V.A.S.; Coirada, F.C.; Boscardin, S.B.; Rosa, D.S. Adjuvants: Classification, Modus Operandi, and Licensing. J. Immunol. Res. 2016, 2016, 1459394.
- Bowen, W.S.; Svrivastava, A.K.; Batra, L.; Barsoumian, H.; Shirwan, H. Current challenges for cancer vaccine adjuvant development. Expert Rev. Vaccines 2018, 17, 207–215.
- Vermaelen, K. Vaccine Strategies to Improve Anti-cancer Cellular Immune Responses. Front. Immunol. 2019, 10, 8.
- Buonsanti, C.; D’Oro, U. Chapter 5—Discovery of Immune Potentiators as Vaccine Adjuvants. In Immunopotentiators in Modern Vaccines, 2nd ed.; Schijns, V.E.J.C., O’Hagan, D.T., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 85–104.
- Olive, C. Pattern recognition receptors: Sentinels in innate immunity and targets of new vaccine adjuvants. Expert Rev. Vaccines 2012, 11, 237–256.