Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Guannan Zhong and Version 3 by Dean Liu.
Lasso peptides are a subclass of ribosomally synthesized and post-translationally modified peptides (RiPPs) and feature the threaded, lariat knot-like topology. The basic post-translational modifications (PTMs) of lasso peptide contain two steps, including the leader peptide removal of the ribosome-derived linear precursor peptide by an ATP-dependent cysteine protease, and the macrolactam cyclization by an ATP-dependent macrolactam synthetase.
- lasso peptides
- post-translational modifications
- biosynthesis
- RiPPs
1. Introduction
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are a family of natural products with remarkable structural variety and functional diversity due to their extensive post-translational modifications (PTMs) [1][2][1,2]. An intriguing member of RiPPs is lasso peptides that consist of an N-terminal macrolactam ring via an isopeptide bond between the α-amine of the first amino acid residue and the carboxylic acid side chain of an aspartate/glutamate located in the 7–9 residues, and a C-terminal tail threads through the macrolactam to form a characteristic lariat topology. The unique knot-like threaded topology endows most lasso peptides with extraordinary stability against heat, proteolysis, and extreme pH conditions, making them distinct from other RiPPs. Diverse physiological functionalities have been reported for lasso peptides, such as antimicrobial, antitumor, antiviral, and receptor antagonistic activities [3][4][5][6][7][3,4,5,6,7].
Generally, at least three gene products are involved in the biosynthesis of lasso peptides: a precursor peptide (A), an ATP-dependent cysteine protease (B), and an ATP-dependent macrolactam synthetase (C). The ribosome-derived linear precursor peptide consists of an N-terminal leader region for the recognition by different PTM enzymes and a C-terminal core region that makes up the structural backbone of lasso peptides. The cysteine protease (B) encompasses a ribosomal recognition element (RRE) domain in the N-terminus to recognize and bind the leader peptide, after which the C-terminal protease domain cleaves the leader peptide to release the leaderless core peptide (prefolded core peptide in Figure 1). In many cases, the N-terminal RRE and the C-terminal protease domains are split into two separate open reading frames (ORFs) termed B1 (RRE) and B2 (cysteine protease), respectively. The nascent core peptide is further delivered to macrolactam cyclase C that uses ATP to activate the side carboxyl of Asp or Glu located at 7–9 as an AMP ester (activated core peptide in Figure 1), followed with the freely N-terminal α-amine attacking the AMP-activated carboxyl group to form an isopeptide bond and achieve the mature lasso peptide (Figure 1). It is very likely that the C-terminal tail of the core peptide is prefolded into the threaded configuration prior to the ring’s closure, otherwise the tail would be excluded by the macrolactam owing to the steric hindrance and unable to form the correct lasso topology. Extra D genes encoding ATP-binding cassette transporters (ABC transporters) are not rare in the biosynthetic gene clusters (BGCs) of lasso peptides and are believed to be responsible for the extracellular transport of mature lasso peptides [3][4][5][6][7][3,4,5,6,7].
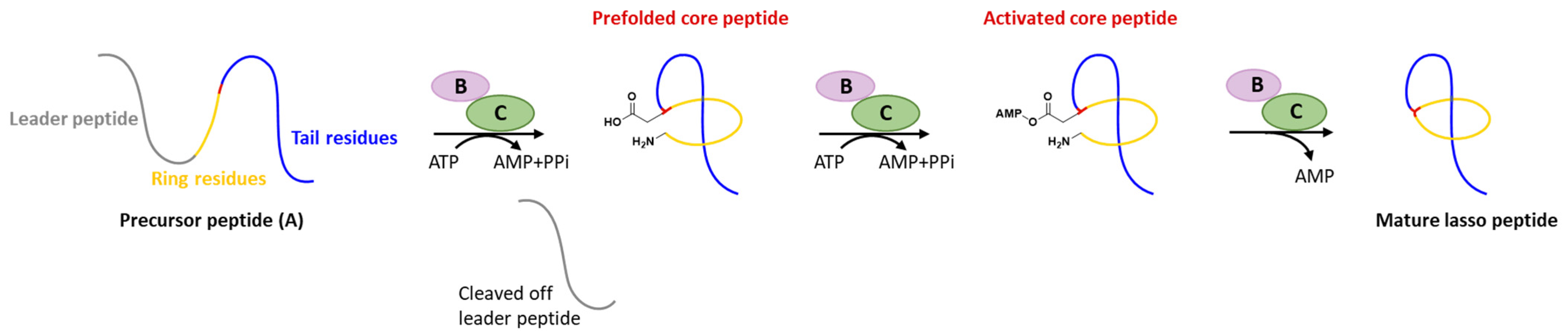
Figure 1. Typical biosynthetic pathway of lasso peptides.
The inexorable progress in genomics, bioinformatics, and chemical analytics greatly facilities lasso peptides discovery during the last decade. In addition to the class-defining modifications as leader peptide excision and core peptide cyclization, a series of unique PTMs including disulfuration, phosphorylation, C-terminal methylation, acetylation, hydroxylation, etc., have been unveiled recently, further increasing the diversity of structures, properties, and complicating the maturation mechanisms.
2. Disulfuration
Disulfide bonds are rare even among all known RiPPs families, and may play an auxiliary role in maintaining the correct configurations, which is curial for biological activities. To the best of theour knowledge, disulfide bonds are only characterized in three classes of RiPPs: glycocins, the post-translationally glycosylated bacteriocins featuring two nested disulfide bonds that stabilize their unique helix–loop–helix structures and sugar moieties on Ser, Thr, or Cys residues [8]; cyclotides, featuring a head-to-tail cyclic peptide backbone with a cystine knot arrangement of three conserved disulfide bonds [9]; and conopeptides, the cone-snail-derived RiPPs containing a high frequency of PTMs involving disulfide bond(s) [10], albeit a few examples in other classes such as lanthipeptides, cyanobactins, sactipeptides, and lasso peptides also contain disulfide(s). Two thiol-disulfide oxidoreductases and a protein-disulfide isomerase (PDI) were reported for the disulfide bond(s) formation in glycocins and cyclotides, respectively [11][12][11,12], while the formation of disulfide bond(s) in conopeptides still remains elusive.3. Phosphorylation
Phosphorylation was the earliest characterized tailoring process in lasso peptides. Paeninodin, originated from firmicute strain Paenibacillus dendritiformis C454, is a class II lasso peptide with a Ser residue in the C-terminus, of which the BGC encodes an additional putative tailoring kinase (PadeK) (Figure 23a) [13][25]. Both unphosphorylated paeninodin and phosphorylated paeninodin were detected in the extract of heterologous expression for paeninodin cluster in Escherichia coli. Deletion of the kinase gene padeK resulted in the production of merely unphosphorylated paeninodin, while restitution of the knocked-out gene by co-expression with another vector-bearing padeK led to restoration of the phosphorylated compound, suggesting the direct link between the function of kinase PadeK and the occurrence of the tailoring phosphorylation process on paeninodin. Precursor peptide PadeA instead of the threaded lasso peptide was verified to be the substrate of kinase PadeK, which specifically modified the hydroxyl group of the C-terminal Ser, the extremely conserved site in the precursor sequences from various lasso peptide BGCs featuring a homologous kinase gene, suggesting the modification step prior to the fundamental maturing process catalyzed by B2 and C proteins (Figure 23b) [13][25]. Owing to the low solubility of PadeK, the homologous kinase ThcoK from another firmicute Thermobacacillus composti KWC4 was chosen instead of PadeK to be characterized in vitro. Replacing padeK with thcoK in the paeninodin heterologous expression system successfully produced the phosphorylated peptide with only minor amounts of unmodified compound, suggesting the feasibility of the hybrid gene cluster. Sequence alignments of lasso peptide-tailoring kinases exposed a conserved His-Lys-Asp-Asp motif. The imperative roles of these four catalytic residues were further demonstrated via site-directed mutations [13][25].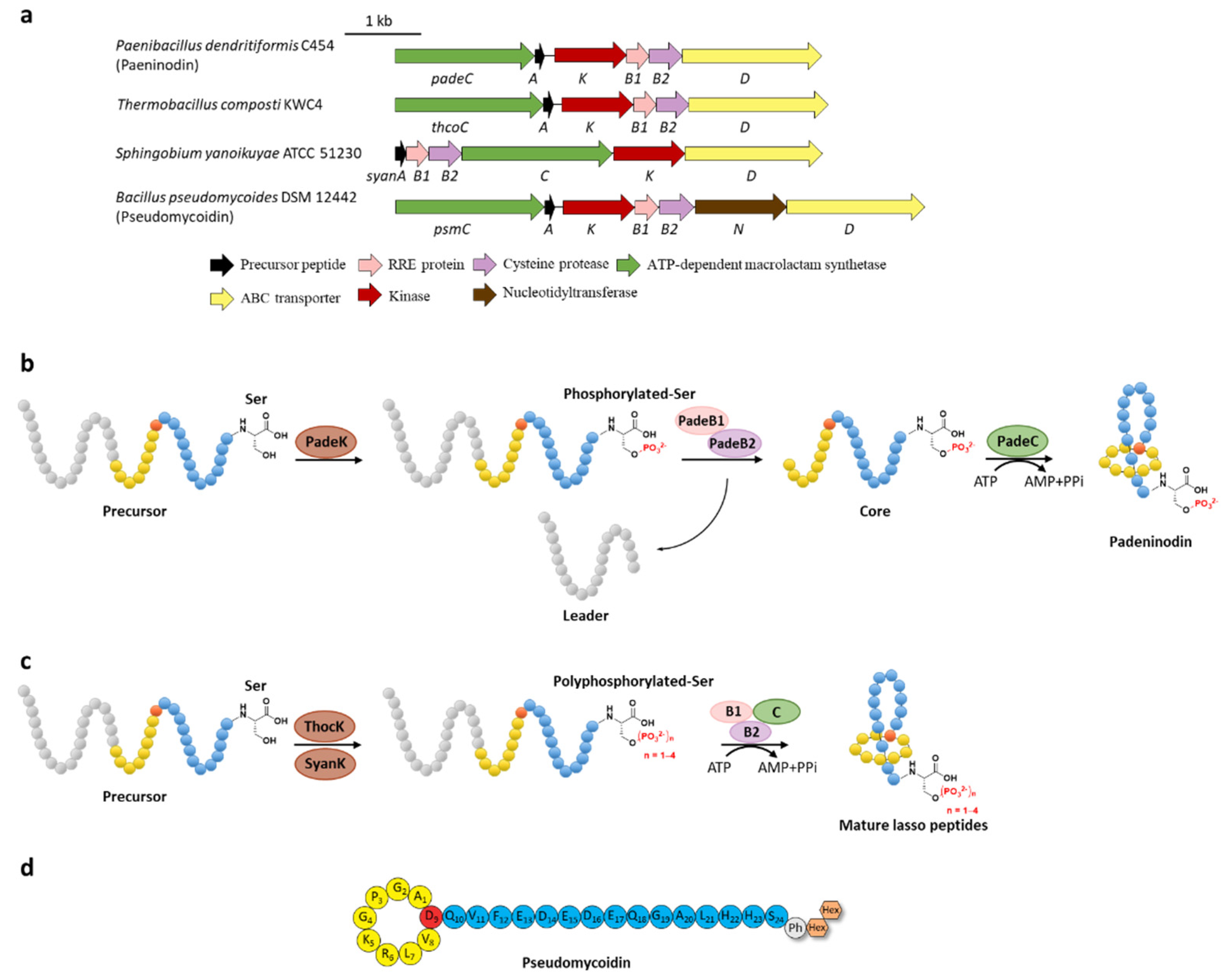 Figure 2. C-terminal phosphorylation of paeninodin and related lasso peptides. (a) The BGCs of phosphorylated lasso peptides. (b) Proposed biosynthetic pathway of paeninodin. The precursor peptide is phosphorylated by PadeK at the C-terminal Ser residue and then maturated by B1, B2, and C proteins to generate paeninodin. (c) Polyphosphorylation of lasso peptides. ThocK and SyanK polyphosphorylated the precursor peptide at the C-terminal Ser residue as well. (d) The structure of pseudomycoidin.
Figure 2. C-terminal phosphorylation of paeninodin and related lasso peptides. (a) The BGCs of phosphorylated lasso peptides. (b) Proposed biosynthetic pathway of paeninodin. The precursor peptide is phosphorylated by PadeK at the C-terminal Ser residue and then maturated by B1, B2, and C proteins to generate paeninodin. (c) Polyphosphorylation of lasso peptides. ThocK and SyanK polyphosphorylated the precursor peptide at the C-terminal Ser residue as well. (d) The structure of pseudomycoidin.
4. Methylation
Methylation is a versatile modification in the biosynthesis of various natural products. Lassomycin discovered from Lentzea kentuckyensis sp. is an absorbing lasso peptide that exhibits outstanding activities against a variety of Mycobacterium tuberculosis strains with minimum inhibitory concentration (MIC) values of 0.8–3 μg/mL and is inactive against symbionts of the human microbiota [14][28]. Although the initial structure elucidation indicated that lassomycin adopted an unthreaded structure [14][28], the subsequent chemical synthesis of this peptide showed that the reported structure was incorrect and a characteristic threaded conformation was essential for its anti-tuberculosis (TB) activity [15][16][29,30]. In addition, lassomycin features a unique methyl ester in the C-terminal carboxyl group, and the putative O-methyltransferase, LasF, from its BGC was considered to be responsible for the C-terminal methylation (Figure 34a) [14][28].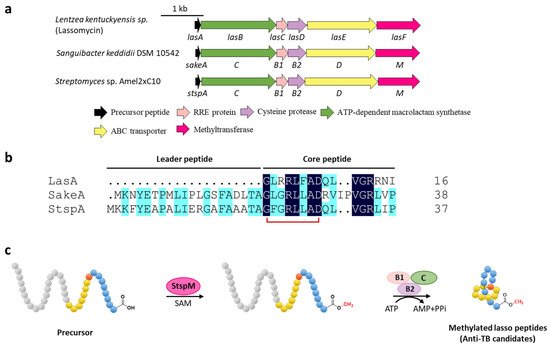
Figure 3. C-terminal methylation of lassomycin and related lasso peptides. (a) The BGCs of methylated lasso peptides. (b) Sequence alignment of precursor peptides. (c) Proposed biosynthetic pathway of C-terminal methylated lasso peptides. StspM methylates the C-terminal carboxyl group of precursor peptide.
5. Acetylation
A novel lasso peptide BGC encoded for albusnodin was found in S. albus DSM 41398 which includes a putative acetyltransferase gene (albT) as well as the canonical genes albA, albB, and albC [17][37]. The only observed heterologous expression product was the threaded, C-terminal Cys truncated albusnodin with an acetyl group attached to the ε-amino group of Lys10 (Figure 45) [17][37]. Sequence alignments showed that Lys10 was highly conserved among precursor peptides in an array of lasso peptide BGCs that resembled the BGC architecture of albusnodin. Heterologous expression of the albusnodin cluster lacking the acetyltransferase gene albT led to no trace of the predicted unacetylated intermediate [17][37], surmising that the acetylation is vital and occurs in the early stage of albusnodin biosynthesis rather than the last step. Moreover, the BGC of the antitumor lasso peptide ulleungdin also contains an acetyltransferase gene in the downstream of B2, yet acetylated ulleungdin was not detected [18][38]. It seems that this acetyltransferase is unrelated to ulleungdin.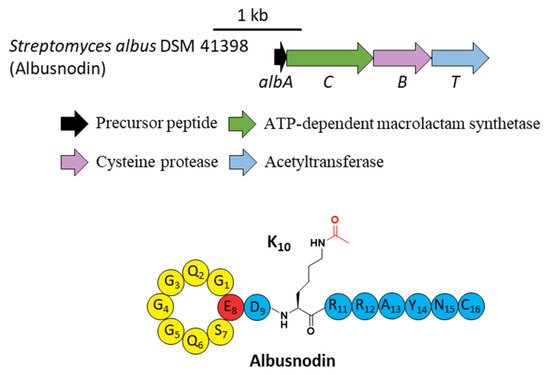
Figure 4. The BGC and structure of albusnodin. The acetyl group attached to the ε-amino of K10 is highlighted in red.
6. Hydroxylation
Lasso peptides RES-701s originally isolated from Streptomyces sp. RE-896 are regarded as selective endothelin type B receptor (ETBR) antagonists. RES-701-2 and RES-701-4 contain a C-terminal 7-hydroxy-tryptophan compared to the unhydroxylated RES-701-1 and RES-701-3 [19][20][21][46,47,48]. Recently, RES-701-3 and RES-701-4 that differed in the hydroxylation of the C-terminal tryptophan residue were rediscovered through genome mining from the marine S. caniferus CA-271066, and their BGC (hereafter termed res) was identified with an additional gene (resE) encoding a hypothetical protein (Figure 6b). Despite lacking any evidence, ResE was proposed for the 7-hydroxylation of the C-terminal tryptophan residue, which remains to be proved in the future [22][49].7. Epimerization
The function of MslH was further validated for the epimerization of the C-terminal l-Trp in vitro [23][51]. The full-length precursor MslA is the most favorite substrate for MslH, as the compared reaction with leaderless core peptide only produced a minor amount of d-Trp. Just like CanB1 in canucin A biosynthesis, MslB1 is also a bifunctional protein that not only assists the proteolysis of leader peptide catalyzed by MslB2, but also remarkably enhances the epimerization activity of MslH. Only about 50% conversion of MslA to epi-MslA was observed, implying that MslH generated an equilibrium mixture of the epimers. Since the C-terminal l-Trp derivative has never been detected in the MS-271 producer, the following MslB2 and MslC maturation processes probably recognize epi-MslA as the sole substrate and drive the equilibrium to d-Trp containing precursor peptide (Figure 67b). Furthermore, MslH could epimerize other aromatic residues such as W21F and W21Y at considerable levels, and chimeric substrates with the sviceucin N-terminal core peptide sequence and the C-terminal “CFW” (Figure 12b), displaying a broad substrate tolerance [23][51].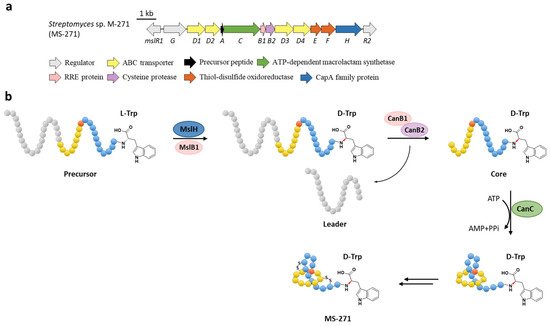
Figure 6. The BGC (a) and proposed biosynthetic pathway (b) of MS-271. MslH epimerizes the C-terminal Trp residue of precursor peptide with the aid of MslB1, similar to the cooperation of CanE and CanB1.
d-amino acids are limited in RiPPs and only a few mechanisms have been verified. For instance, the single radical S-adenosylmethionine (SAM) peptide epimerase PoyD introduces up to 18 d-amino acids in the biosynthesis of polytheonamides [24][52], another radical SAM epimerase YydG epimerizes the formation of a d-Val and d-allo-Ile residues in the biosynthesis of the epipeptide YydF [25][53]. Additionally, d-Ala and d-amino butyric acid (d-Abu) residues are introduced into lanthipeptides by the hydrogenation of 2,3-didehydroalanine (Dha, dehydrated Ser) and 2,3-didehydrobutyrine (Dhb, dehydrated Thr) via different oxidoreductases, including the zinc-dependent dehydrogenases termed LanJA [26][54], the flavin oxidoreductases termed LanJB [27][55], and the F420H2-dependent reductases termed LanJC [28][56]. The characterization of the metallophosphatase superfamily protein MslH provides a novel biosynthetic mechanism for d-amino acids in RiPPs.
8. Citrullination
Citrullination, referring to Arg deimination to produce non-proteinogenic amino acid citrulline (Cit), had never been reported in RiPPs until the lasso peptide citrulassin A was discovered from S. albulus NRRL B-3066 using the Rapid ORF Description and Evaluation Online (RODEO) genome-mining tool. The conversion of Arg9, which is invariable among the citrulassin family, to Cit was certified by in silico analysis of the precursor peptide sequence and nuclear magnetic resonance (NMR) analysis of the maturated citrulassin A (Figure 78b). Heterologous expression of the citrulassin A cluster with ~20 kb upstream and downstream regions only produced des-citrulassin A with unmodified Arg9, suggesting the enzyme responsible for citrulline generation is remotely encoded in the genome [29][22]. Subsequent research revealed that the peptidyl arginine deiminase (PAD) is responsible for deimination of Arg to generate Cit (Figure 78a), as the distantly encoded pad gene was ubiquitous in the genomes of citrulassin producing strains with only one exception, while strains lacking pad correlated to Arg-bearing des-citrulassin production. Heterologous expression of the pad gene in native des-citrulassin D producer (S. katrae NRRL B-16271) resulted in the conversion to deiminated citrulassin D (Figure 78c) [30][57]. Future work is necessary to unveil the timing of deimination during citrulassin biosynthesis.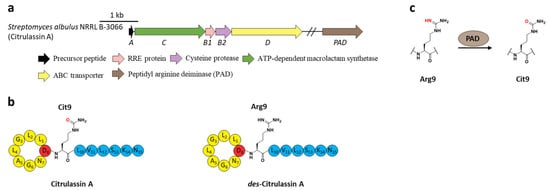
Figure 7. The BGC, structure, and conversion of citrulassin A. (a) The BGC of citrulassin A. The pad gene is distantly encoded in the genome. (b) The structures of citrulassin A and des-citrulassin A. The oxygen atom in Cit9 is highlighted in red. (c) PAD catalyzes the deimination of Arg9 to generate Cit9.
9. Succinimidation
Protein l-isoaspartyl methyltransferases (PIMTs) usually have a crucial role in protein repair, recognizing and repairing abnormal isoaspartate (isoAsp) residues to l-Asp through a SAM-dependent methyl esterification reaction [31][58]. In total, 48 lasso peptide BGCs were uncovered bearing genes annotated as O-methyltransferases that belong to PIMT homologues, and the extremely conserved Asp6 in all the putative precursor peptides suggested that it might be the modification site [32][59]. Heterologous expression of two clusters from actinobacterium Thermobifida cellulosilytica (tce) and firmicute Lihuaxuella thermophila (lih) (Figure 89a) resulted in the discovery of cellulonodin-2 and lihuanodin, featuring an unconventional succinimide moiety (also known as aspartimide) in the macrolactam ring. It was experimentally proved in vitro that TceM and LihM catalyzed the methylation of Asp6 to the corresponding methyl ester, followed with spontaneous nucleophilic attack of the adjacent Thr7 amino group to form a stable succinimide moiety without further hydrolyzation. Notably, TceM and LihM carried out dehydration on Asp instead of isoAsp, which is in stark contrast to canonical PIMTs. In addition, both TceM and LihM only recognized the threaded lasso peptides rather than linear precursors or isopeptide-bonded rings (Figure 89b) [32][59]. The functions of TceM and LihM are distinct from the previously reported PIMT OlvSA involved in the biosynthesis of lanthipeptide OlvA (BCSA), since the OlvSA catalytic succinimide group was followed with non-enzymatic hydrolysis to either Asp or isoAsp and this process was reversible as isoAsp could be recognized by OlvSA as well to regenerate succinimide [33][60].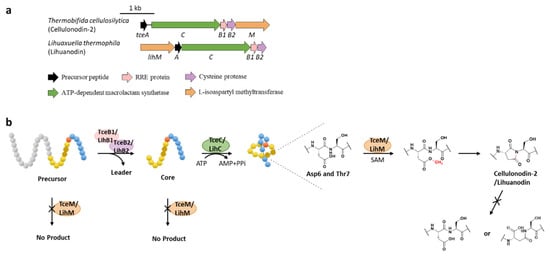
Figure 8. The BGCs (a) and biosynthetic pathways (b) of cellulonodin-2 and lihuanodin. Zoomed-in images of the Asp6 and Thr7 residues in the threaded lasso peptides are provided for further illustration. Both TceM and LihM could only recognize the threaded lasso peptides as substrates.