Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jessie Wu and Version 1 by Kajal Gupta.
Toll-like receptors (TLRs) are typical transmembrane proteins, which are essential pattern recognition receptors in mediating the effects of innate immunity. TLRs recognize structurally conserved molecules derived from microbes and damage-associated molecular pattern molecules that play an important role in inflammation.
- cancer
- Toll-like receptors
- immunotherapy
1. TLoll like Receptor Localization and Recognition of Microbial Ligands
Toll like receptors (TLRs) are involved in the recognition of microbial exogenously and endogenously derived molecular patterns. This occurs at the plasma membrane and at intracellular compartments, respectively, and thus TLR ligands can be either exogenous or host-derived. TLRs 1, 2, 4, 5, and 6 are located primarily in the plasma membrane, where they interact with components of microbial pathogens that exogenously encounter the cell. In contrast, TLRs 3, 7, 8, and 9 are situated in the membranes of endosomes and lysosomes, where they interact with components that are endogenous. Figure 1 depicts in detail the location of specific TLRs and their respective, best-characterized ligands.
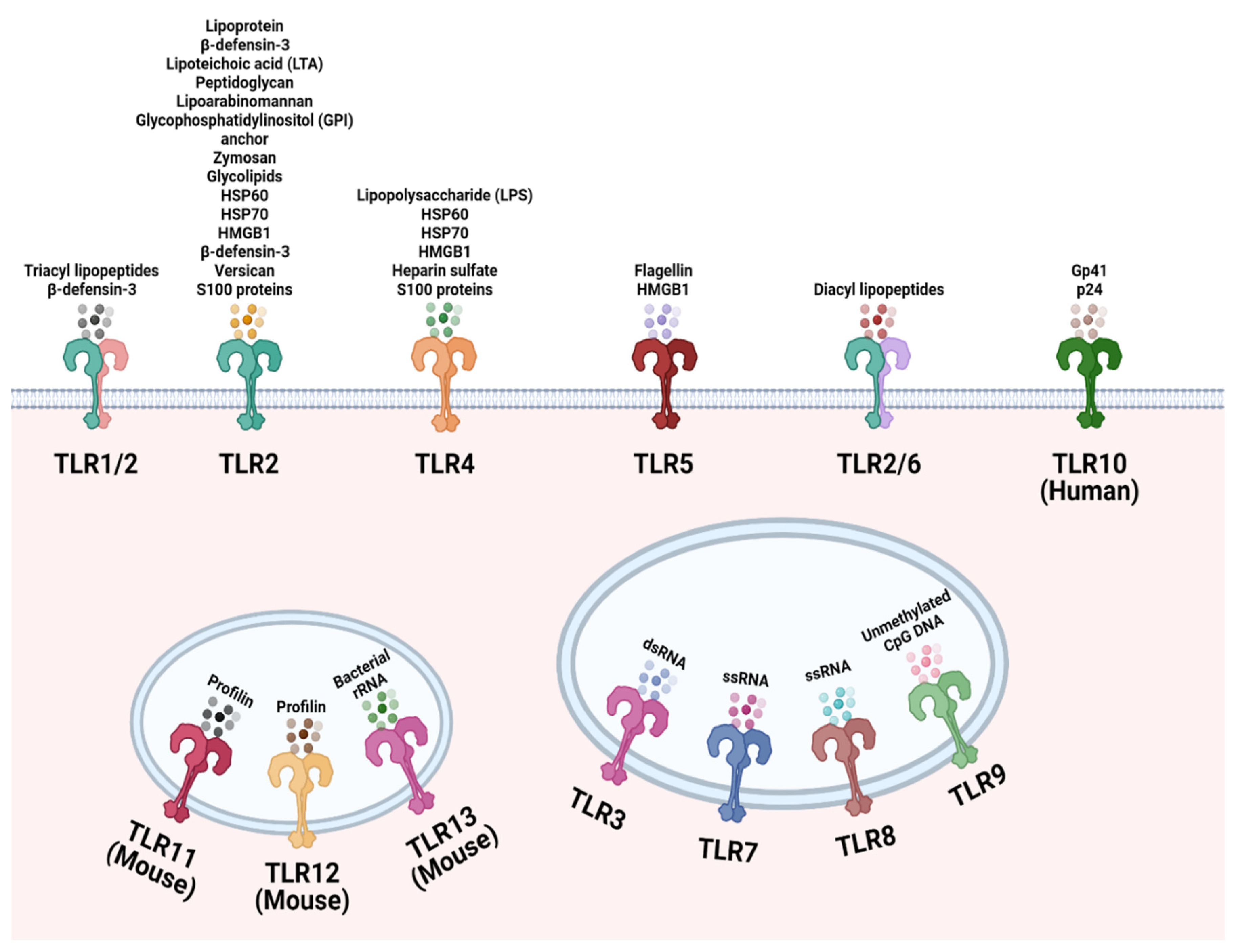
Figure 1. Cellular localization of the Toll-like receptor (TLR) family. TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10 are localized to the cell surface to recognize common microbial structural components and endogenous ligands. TLR3, TLR7, TLR8, TLR9, TLR12 and TLR13 are located on endosomes to sense microbial nucleic acids that have entered the cell. TLR11 and TLR12 are localized to endosomes to recognize Toxoplasma gondii derived profilin. Created with Biorender.com.
2. Microbial Derived TLoll like Receptor Agonists and Their Role in Cancer Immunotherapy
Harnessing the host response to infection has been utilized to target cancer for centuries [81,82][1][2]. Microbial sensing through TLRs initiates a signaling cascade that induces pro-inflammatory responses. The Toll-like receptor signaling pathway plays a crucial role in host immune defenses against numerous diseases and has been identified as an immunotherapeutic target against various types of cancer. New avenues to combat cancers involves the regulation of the host’s innate immune system using agonists, which can bind to a variety of TLRs. These agonists can be used in combination with other cancer therapies, like chemotherapy and radiotherapy, to provide a broader spectrum of protection.
3. Bacterial-Derived TLoll like Receptor Agonists
Many of the pivotal studies in the field of bacterial-based cancer immunotherapy (BBCT) were pioneered by 19th century clinician–scientist Dr. William Coley [81,82][1][2]. Dr. Coley utilized a combination of live Streptococcus pyogenes and Serratia marcescens to treat patients with inoperable sarcomas [81][1]. In the patients with the most dramatic tumor regression, Dr. Coley noted that an erysipelas infection and subsequent fever had been induced in these patients. Dr. Coley’s observations were likely a result of robust immune system activation from Coley’s bacterial-based toxins, thus implicating a link between response to infection and tumor eradication that would serve as the basis for modern cancer immunotherapies. Though unbeknownst to Dr. Coley, a likely mediator in his patients’ responses was innate microbial recognition through Toll-like receptors [82,83][2][3].
Molecular triggers responsible for remissions from Coley’s toxins are TLR ligands that target patter recognition receptors (PRRs) [84][4]. PRRs share the ability to recognize relatively conserved microbial components, which are generally referred to as microbe- or pathogen-associated molecular patterns (MAMPs or PAMPs), as well as endogenous danger signals commonly known as damage-associated molecular patterns (DAMPs). Common TLR-activating MAMPs include viral and bacterial nucleic acids (which can signal through TLR3, TLR7, TLR8, or TLR9), flagellin (a TLR5 agonist), as well as lipopolysaccharide (LPS), lipoteichoic acid, and mannans (which signal through TLR2 or TLR4). Endogenous nucleic acids and the nuclear non-histone protein high mobility group box 1 (HGMB1) are prototypic TLR-activating DAMPs.
Due to their role in self/nonself-differentiation [85][5] and their ability to induce antigen presenting cell (APC) maturation, TLR agonists are considered promising adjuvant candidates [86][6]. In fact, a number of TLR agonists, including Pam3CSK4, Pam2CSK4, MPLA (a LPS derivative), CpG, PolyI:C, and flagellin, are currently being tested as cancer agonists [87][7].
Roberts et al. recently showed that an attenuated strain of Clostridium novyi efficiently decreased tumor size in rat and dog cancer models in addition to one sarcoma patient [88][8]. This treatment is well-targeted as spores of Clostridium novyi germinate selectively within the hypoxic regions of cancerous tissue and induce immune responses likely via TLR activation [89][9].
4. Viral-Derived TLoll like Receptor Agonists
Oncolytic viruses (OVs) preferentially target tumor cells and activate antitumor immunity while limiting pathogenicity, thus they have emerged as a promising tool for viral-based cancer therapies [90][10]. Though additional studies must be conducted to further characterize the TLR contribution to antitumor responses from oncolytic virotherapy, OVs possess several TLR-stimulating moieties that can contribute to activation of host immune response at the tumor site. Intratumoral treatment of murine glioma tumors with oncolytic adenovirus Delta-24-RGD significantly reduced tumor growth compared to PBS-treated tumors [91][11]. Delta-24-RGD treatment was demonstrated to remodel the tumor microenvironment, predominantly through enhanced CD8+ and CD4+ T cell infiltration in the tumor [91][11]. Immune-mediated targeting of the tumor is likely a result of TLR9-medated T cell proliferation as well as maturation of APCs, following the TLR9 recognition of double-stranded adenoviral DNA [92,93,94][12][13][14].
Enhancing the intrinsic TLR stimulating properties of oncolytic adenoviruses has been another strategy for targeting TLR-mediated immune responses. Ad5D24-CpG, an oncolytic adenovirus genetically manipulated to express TLR9 stimulating CpG islands, significantly controlled tumor growth compared to CpG unenhanced Ad5D24 treated tumors [95][15]. Antitumor response from Ad5D24-CpG treatment was determined to be highly reliant on TLR9-mediated NK cell activation, leading to the effective killing of tumor cells [95][15]. Because stimulation of TLRs expressed on NK cells enhances release of cytotoxic granules and cytokine production, in this context it is suggested that the CpG insertion in Ad5D24 enhances these NK cell properties, facilitating more robust antitumor responses [96,97][16][17].
TLRs have also been implicated in the usage of non-oncolytic viruses for viral-based therapeutics. The intratumoral administration of heat-inactivated influenza but not active influenza virus was found to drastically inhibit tumor growth in a B16 melanoma model [98][18]. This response from heat-inactivated influenza treatment was determined to be dependent on increased cross-presenting CD8+ dendritic cell infiltration in the tumor [98][18]. Paralleling the in vivo findings, heat-inactivated influenza was observed to more potently stimulate TLR7 compared to active influenza virus, determined by using TLR7 reporter cells [98][18]. Given that the recognition of TLR ligands can enhance antigen cross presentation in DCs, it is likely that heat-inactivated influenza more potently engaged TLR7, leading to an increase in cross-presenting DCs within the tumor and the activation of CD8+ T cells and thereby reduced tumor progression [99,100,101][19][20][21].
5. TLoll like Receptor Signaling in Cancer
5.1. Effects of Tumor-Promoting TLoll like Receptor Signaling
TLRs have been associated with tumorigenesis, as they can activate multiple cancer-associated signaling pathways. To date, TLRs have been recognized to transduce signals through NF-κB, PI3k-Akt, and MAPK-ERK to advance cancer. NF-κB is recognized as the canonical signaling target upon TLR activation. When activated, NF-κB plays a role in several cellular functions, including cell proliferation, pro-inflammatory cytokine production, and cell survival/apoptosis, as described in Figure 2 [131][22]. As a result of regulating many cellular processes, the NF-κB pathway is an optimal target for aberrant, pro-tumorigenic signaling [132][23]. Through the LPS stimulation of TLR4, NF-κB activation enhanced the proliferation of gastric cancer cell lines BGC-823 and SGC-7901 [131,133][22][24]. In similar form, NF-κB activation via TLR4 resulted in apoptosis evasion in lung and head and neck cancer, with the addition of TNF-related apoptosis-inducing ligand (TRAIL) [134,135][25][26].
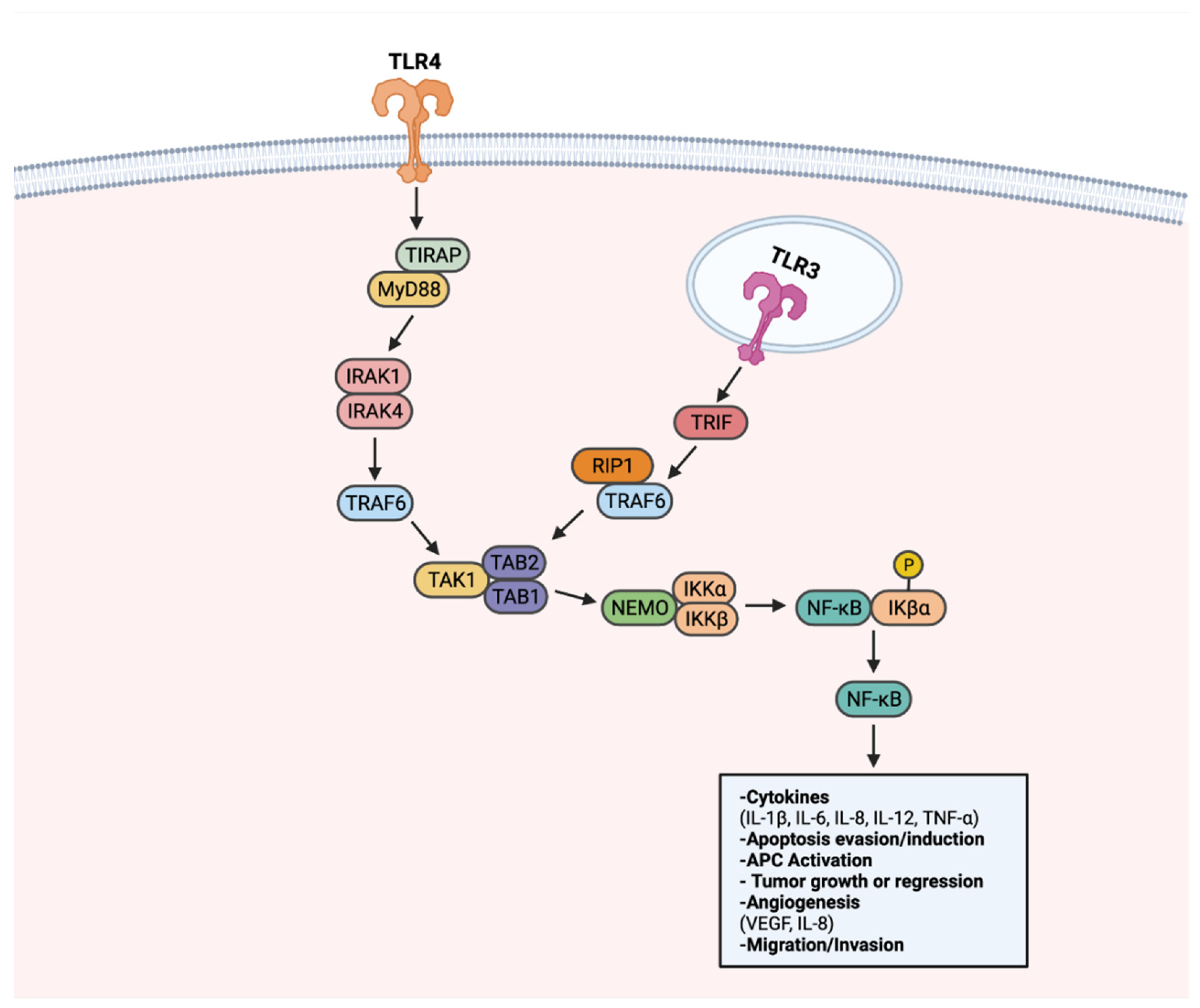
Figure 2. TLR-NF-κB signaling in progression or regression of cancer. Upon recognition of a ligand, TLRs transduce signals through a MyD88-dependent signaling pathway, or alternatively a MyD88-independent TRIF-activating pathway. Upon activation of the MyD88-dependent pathway, adapter molecule TIRAP transduces signals to MyD88. MyD88 activates IRAK1 and IRAK4. Activated IRAK1 and IRAK4 lead to activation of TRAF6. TRAF6 activates the TAK1 complex. The TAK1 complex activates the IKK complex comprised of NEMO, IKKα, and IKKβ. IKK complex activation leads to phosphorylation of IKβα, a protein responsible for sequestering NF-κB to the cytoplasm. Once activated, NF-κB translocates to the nucleus to activate genes that can promote or inhibit tumorigenesis. NF-κB also can be activated through TRIF, notably through TLR3. TRIF activation results in RIP1 and TRAF6 activation. Through RIP1 and TRAF6, the TAK1 complex is activated. Following TAK1 complex activation, subsequent steps in NF-κB signaling are shared between the two pathways. Created with Biorender.com.
Tumorigenic TLR signaling extends beyond the canonical NF-κB pathway. Adaptor protein BCAP has been recognized as a link to TLR activation and downstream PI3k-Akt signaling [136,137][27][28]. PI3K-Akt signaling regulates cell proliferation, cell growth, and survival, thus is frequently upregulated in many cancers [138][29] (Figure 3). TLR7 activation was found to perpetuate pancreatic cancer progression through upregulated PI3K/Akt signaling. Consequently, an upregulation of downstream signaling targets was also observed, including antiapoptotic and pro-proliferative genes Bcl-xL and c-Myc [64,134][25][30]. Apoptosis resistance in PCI-30 cells has been shown to be mediated by TLR4-PI3K/Akt signaling [135][26]. Enhanced angiogenesis has also been described as a product of PI3K-Akt signaling, particularly in a cancer context. TLR4 stimulation in PANC-1 cells has increased vasculature formation and proliferation through PI3K/Akt dependent signaling [138][29].
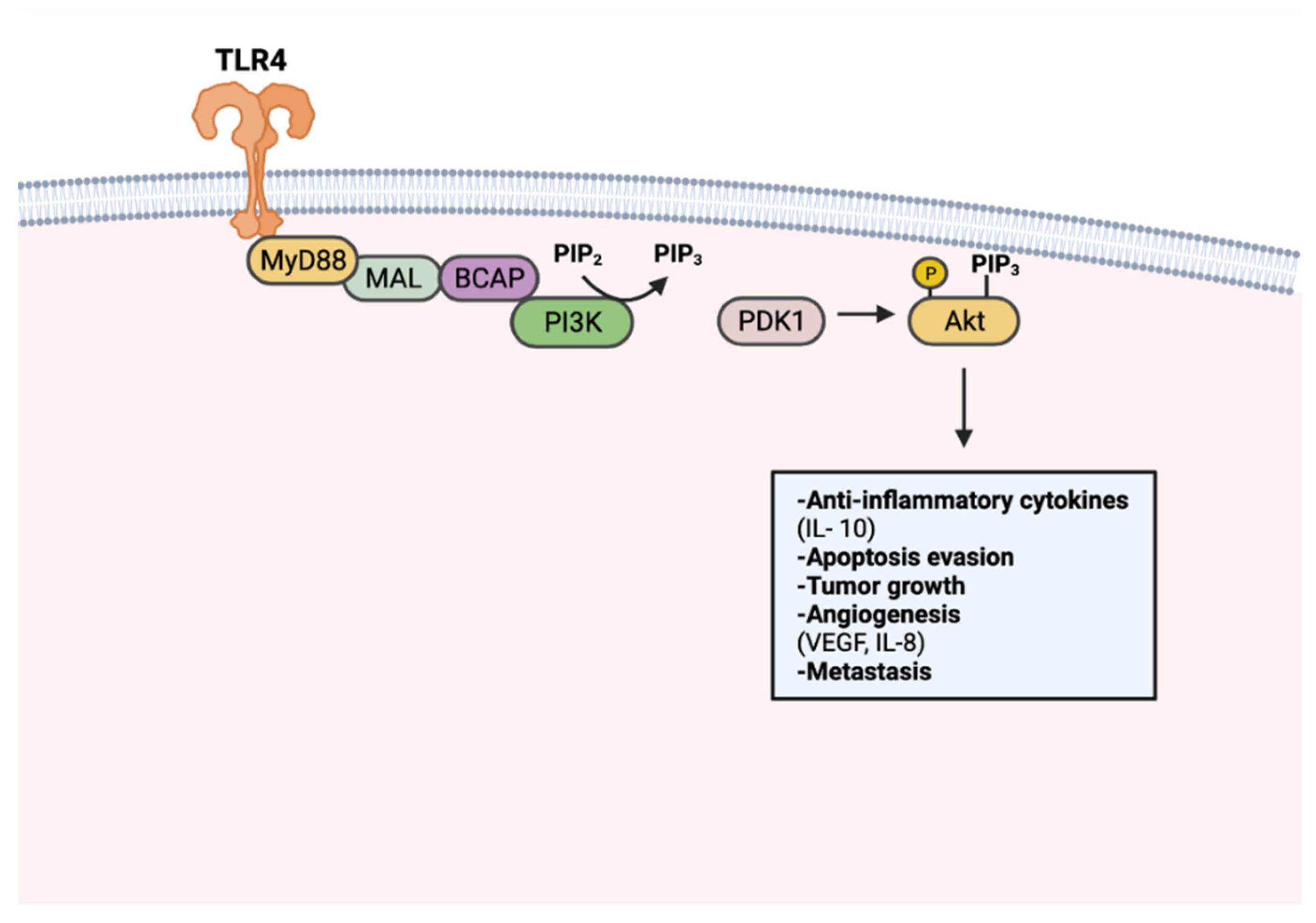
Figure 3. TLR-mediated PI3k-Akt signaling in tumorigenesis. PI3k is activated upon TLR stimulation through MyD88, MAL, and BCAP. Activated PI3k converts phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol (3,4,5)-trisphosphate (PIP3). Akt activation occurs through PIP3-facilitated recruitment to the plasma membrane and phosphorylation by PDK1. Activated Akt promotes tumor progression through anti-inflammatory cytokine production, apoptosis resistance, tumor growth, angiogenesis, and metastasis. Created with Biorender.com.
Aberrant MAPK/ERK signaling has been heavily implicated in approximately a third of all human cancers [139][31]. Through activation of TAK1, TLR stimulation can activate the MAPK/ERK pathway to regulate growth, cell survival, and metastasis [140][32] (Figure 4). LPS-induced TLR4 stimulation in A549 and H1299 cells was found to promote secretion of pro-angiogenic factors VEGF and IL-8 in a p38 MAPK dependent manner [141][33]. MAPK/ERK signaling utilizes transcription regulation to promote tumoral immune evasion [140][32]. Endogenous activation of TLR2 on glioma-associated microglia was observed to downregulate MHC class II expression and impede antigen presentation in a TLR2-ERK1/2-dependent manner [142,143][34][35]. As a result, proliferation and activation of glioma targeting CD4+ T cells was hindered.
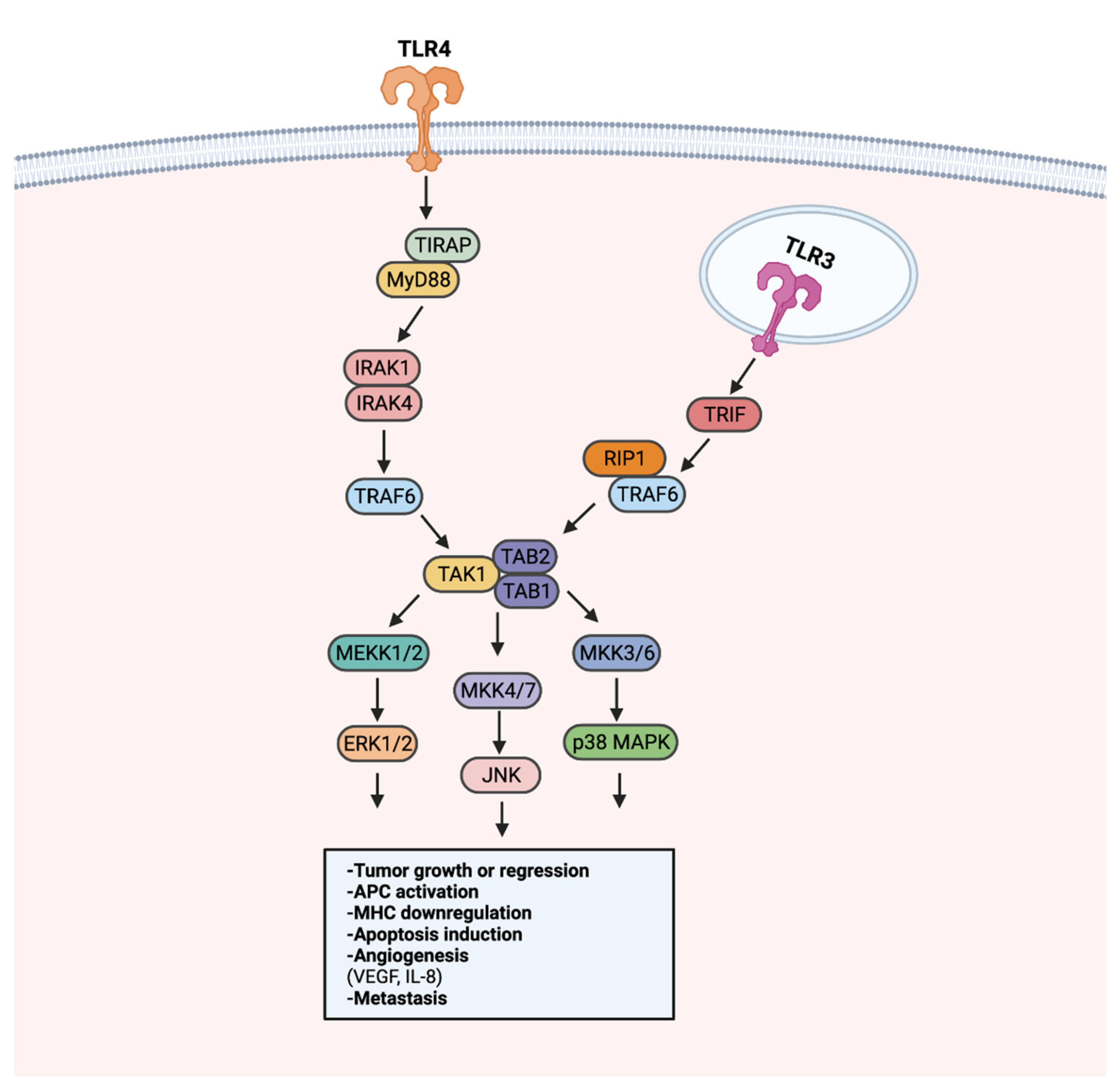
Figure 4. TLR-MAPK/ERK signaling in the progression or regression of cancer. MAPK/ERK pathway activation can occur through MyD88-dependent or MyD88-independent TRIF signaling. The MyD88-dependent and -independent signaling converges in the activation of the TAK1 complex. Following the activation of the TAK1 complex, MEKK1/2, MKK4/7, and MKK3/6 are activated. MEKK1/2, MKK4/7, and MKK3/6 activation leads to the activation of ERK1/2, JNK, and p38 MAPK, respectively. Signaling from ERK1/2, JNK, and p38 MAPK can promote tumor growth or inhibition, APC modulation, angiogenesis, and metastasis. Created with Biorender.com.
5.2. Effects of Anti-Tumor TLoll like Receptor Signaling
The fate of pro- or anti-tumor TLR signaling seems to be largely context-dependent, with considerations being the cancer type, the ligand activating the TLR, and the TLR itself. Tumor-rejecting TLR signaling utilizes several of the same pathways that perpetuate tumor growth, including NF-κB. TLR-mediated NF-κB signaling has been shown to induce IL-1β, IL-6, and TNF-α production in breast and bladder cancer models [26,144][36][37]. Targeting MAPK/ERK signaling has also been reported to promote anti-tumor responses. Dendritic cells stimulated with TLR3 and TLR7 agonists were found to upregulate ERK signaling, likely contributing to the enhanced dendritic cell activation and anti-tumor T-cell responses observed in CT26 tumors [145][38].
Apoptosis-induction in cancer cells appears to be a primary target for anti-tumor TLR signaling. Unsurprisingly, modulating NF-κB signaling has been of interest to promote cancer cell apoptosis [131][22]. The poly(I:C) stimulation of TLR3 on PCI-15B cells was reported to enhance apoptosis through sustained NF-κB inactivation [146][39]. As a result, inactive NF-κB is likely unable to activate downstream anti-apoptotic genes, such as BCL-2 [131][22]. In contrast, TLR3-mediated NF-κB activation was found to be required for apoptosis in poly(I:C)-treated CAMA-1 cells [22][40]. It appears that the activation or inactivation of NF-κB to promote apoptosis is dependent on the cancer type and additional factors that may be driving the cancer.
Beyond NF-κB, the stimulation of TLRs can activate a type I interferon (IFN) signaling pathway to promote anti-tumor responses (Figure 5). Because IFN signaling can occur to induce apoptosis in infected cells, it has been recognized as a beneficial tool for initiating apoptosis in malignant cells. Treating LNCaP cells with a TLR3 agonist was found to promote apoptosis in an IRF-3 signaling-dependent manner. Through TLR–IFN signaling, pro-apoptotic protein Noxa was subsequently upregulated downstream, a likely contributor to the increased cancer cell death [147][41]. Apoptosis mediated by IFN signaling extends to other cancer types. In a non-muscle invasive bladder cancer model, TLR4 activation with P-MAPA was shown to enhance IFN signaling. TLR4-mediated IFN signaling resulted in increased apoptosis and iNOS expression, an enzyme responsible for nitric oxide (NO) synthesis [144][37]. Increased intracellular NO concentrations have been shown to induce apoptosis [148][42], thus it is likely that the apoptosis of bladder cancer tumors occurs through a TLR4–IFN–iNOS axis [149][43].
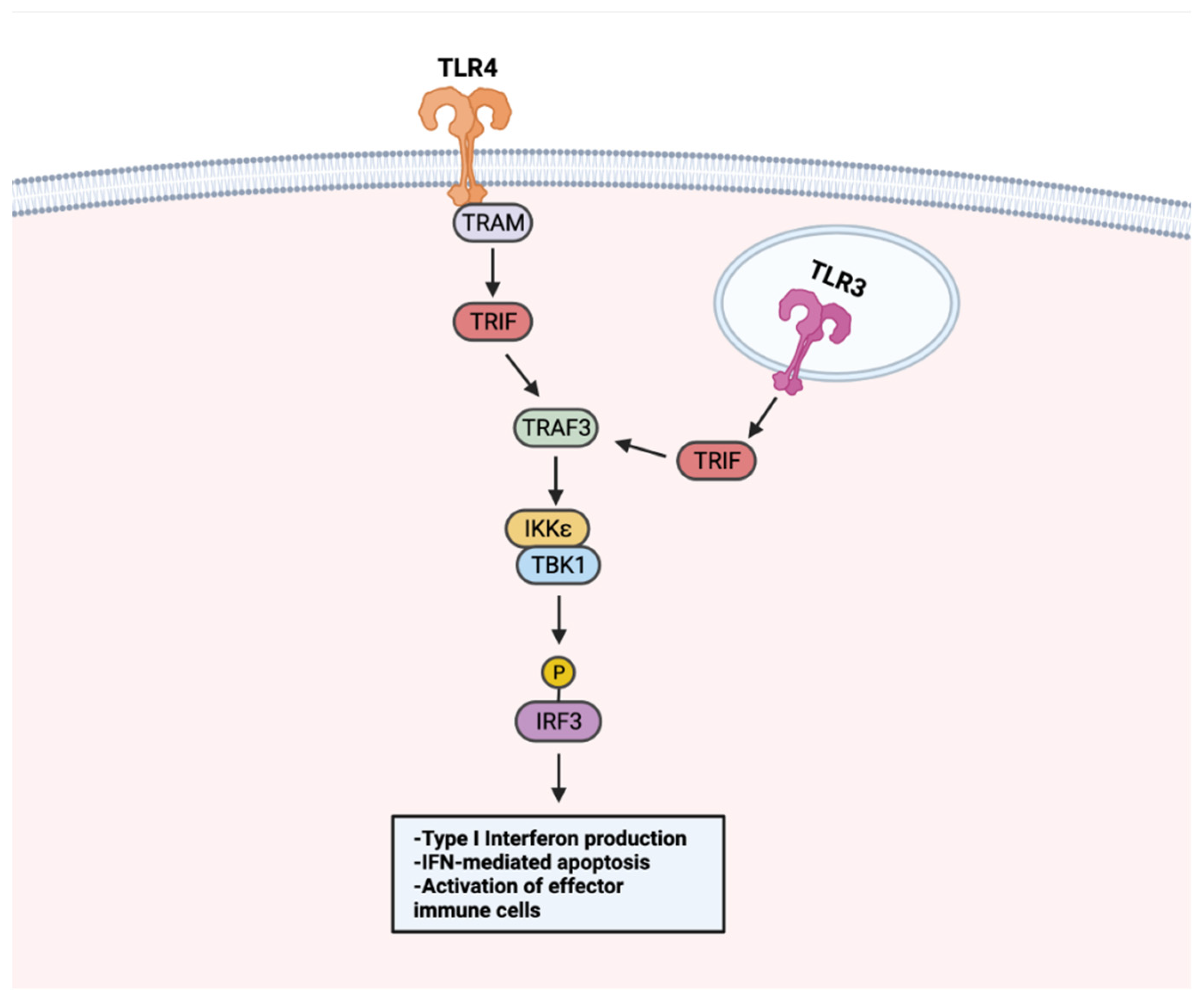
Figure 5. TLR–IFN signaling in anti-tumor responses. TLRs use a MyD88-independent pathway for IFN signaling. Upon TLR stimulation, membrane-localized TLRs (i.e., TLR4) activate TRIF through adaptor protein TRAM. TRIF activates TRAF3, which activates the TBK1–IKKε complex. The TBK–IKKε complex phosphorylates IRF3. Endosome-localized TLR3 activates TRIF directly. Following TRIF activation, TRAF3 is activated and shares downstream IFN signaling steps with membrane-localized TLRs. TLR–IFN signaling induces type I IFN production, apoptosis, and activation of immune cells. Created with Biorender.com.
References
- Coley, W.B. The Treatment of Inoperable Sarcoma by Bacterial Toxins (the Mixed Toxins of the Streptococcus erysipelas and the Bacillus prodigiosus). Proc. R. Soc. Med. 1910, 3, 1–48.
- Coley, W.B. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. Am. J. Med. Sci. 1893, 105, 487.
- Gupta, K.H.; Nowicki, C.; Giurini, E.F.; Marzo, A.L.; Zloza, A. Bacterial-Based Cancer Therapy (BBCT): Recent Advances, Current Challenges, and Future Prospects for Cancer Immunotherapy. Vaccines 2021, 9, 1497.
- Orange, M.; Reuter, U.; Hobohm, U. Coley’s Lessons Remembered: Augmenting Mistletoe Therapy. Integr. Cancer Ther. 2016, 15, 502–511.
- Hobohm, U.; Stanford, J.L.; Grange, J.M. Pathogen-associated molecular pattern in cancer immunotherapy. Crit. Rev. Immunol. 2008, 28, 95–107.
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216.
- Bendelac, A.; Medzhitov, R. Adjuvants of immunity: Harnessing innate immunity to promote adaptive immunity. J. Exp. Med. 2002, 195, F19–F23.
- Roberts, N.J.; Zhang, L.; Janku, F.; Collins, A.; Bai, R.Y.; Staedtke, V.; Rusk, A.W.; Tung, D.; Miller, M.; Roix, J.; et al. Intratumoral injection of Clostridium novyi-NT spores induces antitumor responses. Sci. Transl. Med. 2014, 6, 249ra111.
- Agrawal, N.; Bettegowda, C.; Cheong, I.; Geschwind, J.F.; Drake, C.G.; Hipkiss, E.L.; Tatsumi, M.; Dang, L.H.; Diaz, L.A., Jr.; Pomper, M.; et al. Bacteriolytic therapy can generate a potent immune response against experimental tumors. Proc. Natl. Acad. Sci. USA 2004, 101, 15172–15177.
- Macedo, N.; Miller, D.M.; Haq, R.; Kaufman, H.L. Clinical landscape of oncolytic virus research in 2020. J. Immunother. Cancer 2020, 8, e001486.
- Martínez-Vélez, N.; Garcia-Moure, M.; Marigil, M.; González-Huarriz, M.; Puigdelloses, M.; Gallego Pérez-Larraya, J.; Zalacaín, M.; Marrodán, L.; Varela-Guruceaga, M.; Laspidea, V.; et al. The oncolytic virus Delta-24-RGD elicits an antitumor effect in pediatric glioma and DIPG mouse models. Nat. Commun. 2019, 10, 2235.
- Krieg, A.M. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene 2008, 27, 161–167.
- Rahman, A.H.; Taylor, D.K.; Turka, L.A. The contribution of direct TLR signaling to T cell responses. Immunol. Res. 2009, 45, 25–36.
- Kabelitz, D. Expression and function of Toll-like receptors in T lymphocytes. Curr. Opin. Immunol. 2007, 19, 39–45.
- Cerullo, V.; Diaconu, I.; Romano, V.; Hirvinen, M.; Ugolini, M.; Escutenaire, S.; Holm, S.L.; Kipar, A.; Kanerva, A.; Hemminki, A. An oncolytic adenovirus enhanced for toll-like receptor 9 stimulation increases antitumor immune responses and tumor clearance. Mol. Ther. 2012, 20, 2076–2086.
- Noh, J.Y.; Yoon, S.R.; Kim, T.D.; Choi, I.; Jung, H. Toll-Like Receptors in Natural Killer Cells and Their Application for Immunotherapy. J. Immunol. Res. 2020, 2020, 2045860.
- Adib-Conquy, M.; Scott-Algara, D.; Cavaillon, J.M.; Souza-Fonseca-Guimaraes, F. TLR-mediated activation of NK cells and their role in bacterial/viral immune responses in mammals. Immunol. Cell Biol. 2014, 92, 256–262.
- Newman, J.H.; Chesson, C.B.; Herzog, N.L.; Bommareddy, P.K.; Aspromonte, S.M.; Pepe, R.; Estupinian, R.; Aboelatta, M.M.; Buddhadev, S.; Tarabichi, S.; et al. Intratumoral injection of the seasonal flu shot converts immunologically cold tumors to hot and serves as an immunotherapy for cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 1119–1128.
- Datta, S.K.; Redecke, V.; Prilliman, K.R.; Takabayashi, K.; Corr, M.; Tallant, T.; DiDonato, J.; Dziarski, R.; Akira, S.; Schoenberger, S.P.; et al. A subset of Toll-like receptor ligands induces cross-presentation by bone marrow-derived dendritic cells. J. Immunol. 2003, 170, 4102–4110.
- Crespo, M.I.; Zacca, E.R.; Núñez, N.G.; Ranocchia, R.P.; Maccioni, M.; Maletto, B.A.; Pistoresi-Palencia, M.C.; Morón, G. TLR7 triggering with polyuridylic acid promotes cross-presentation in CD8α+ conventional dendritic cells by enhancing antigen preservation and MHC class I antigen permanence on the dendritic cell surface. J. Immunol. 2013, 190, 948–960.
- Oh, J.Z.; Kurche, J.S.; Burchill, M.A.; Kedl, R.M. TLR7 enables cross-presentation by multiple dendritic cell subsets through a type I IFN-dependent pathway. Blood 2011, 118, 3028–3038.
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023.
- Park, M.H.; Hong, J.T. Roles of NF-κB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15.
- Yuan, X.; Zhou, Y.; Wang, W.; Li, J.; Xie, G.; Zhao, Y.; Xu, D.; Shen, L. Activation of TLR4 signaling promotes gastric cancer progression by inducing mitochondrial ROS production. Cell Death Dis. 2013, 4, e794.
- He, W.; Liu, Q.; Wang, L.; Chen, W.; Li, N.; Cao, X. Corrigendum to “TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance” . Mol. Immunol. 2020, 122, 232–234.
- Szczepanski, M.J.; Czystowska, M.; Szajnik, M.; Harasymczuk, M.; Boyiadzis, M.; Kruk-Zagajewska, A.; Szyfter, W.; Zeromski, J.; Whiteside, T.L. Triggering of Toll-like receptor 4 expressed on human head and neck squamous cell carcinoma promotes tumor development and protects the tumor from immune attack. Cancer Res. 2009, 69, 3105–3113.
- Luo, L.; Lucas, R.M.; Liu, L.; Stow, J.L. Signalling, sorting and scaffolding adaptors for Toll-like receptors. J. Cell Sci. 2019, 133, 239194.
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88.
- Sun, Y.; Wu, C.; Ma, J.; Yang, Y.; Man, X.; Wu, H.; Li, S. Toll-like receptor 4 promotes angiogenesis in pancreatic cancer via PI3K/AKT signaling. Exp. Cell Res. 2016, 347, 274–282.
- Ochi, A.; Graffeo, C.S.; Zambirinis, C.P.; Rehman, A.; Hackman, M.; Fallon, N.; Barilla, R.M.; Henning, J.R.; Jamal, M.; Rao, R.; et al. Toll-like receptor 7 regulates pancreatic carcinogenesis in mice and humans. J. Clin. Investig. 2012, 122, 4118–4129.
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290.
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 103–119.
- He, W.; Liu, Q.; Wang, L.; Chen, W.; Li, N.; Cao, X. TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol. Immunol. 2007, 44, 2850–2859.
- Kong, C.; Yan, X.; Zhu, Y.; Zhu, H.; Luo, Y.; Liu, P.; Ferrandon, S.; Kalady, M.F.; Gao, R.; He, J.; et al. Fusobacterium Nucleatum Promotes the Development of Colorectal Cancer by Activating a Cytochrome P450/Epoxyoctadecenoic Acid Axis via TLR4/Keap1/NRF2 Signaling. Cancer Res. 2021, 81, 4485–4498.
- Qian, J.; Luo, F.; Yang, J.; Liu, J.; Liu, R.; Wang, L.; Wang, C.; Deng, Y.; Lu, Z.; Wang, Y.; et al. TLR2 Promotes Glioma Immune Evasion by Downregulating MHC Class II Molecules in Microglia. Cancer Immunol. Res. 2018, 6, 1220–1233.
- Cai, Z.; Sanchez, A.; Shi, Z.; Zhang, T.; Liu, M.; Zhang, D. Activation of Toll-like receptor 5 on breast cancer cells by flagellin suppresses cell proliferation and tumor growth. Cancer Res. 2011, 71, 2466–2475.
- Garcia, P.V.; Seiva, F.R.; Carniato, A.P.; de Mello Júnior, W.; Duran, N.; Macedo, A.M.; de Oliveira, A.G.; Romih, R.; Nunes Ida, S.; Nunes Oda, S.; et al. Increased toll-like receptors and p53 levels regulate apoptosis and angiogenesis in non-muscle invasive bladder cancer: Mechanism of action of P-MAPA biological response modifier. BMC Cancer 2016, 16, 422.
- Hotz, C.; Treinies, M.; Mottas, I.; Rötzer, L.C.; Oberson, A.; Spagnuolo, L.; Perdicchio, M.; Spinetti, T.; Herbst, T.; Bourquin, C. Reprogramming of TLR7 signaling enhances antitumor NK and cytotoxic T cell responses. Oncoimmunology 2016, 5, e1232219.
- Umemura, N.; Zhu, J.; Mburu, Y.K.; Forero, A.; Hsieh, P.N.; Muthuswamy, R.; Kalinski, P.; Ferris, R.L.; Sarkar, S.N. Defective NF-κB signaling in metastatic head and neck cancer cells leads to enhanced apoptosis by double-stranded RNA. Cancer Res. 2012, 72, 45–55.
- Salaun, B.; Coste, I.; Rissoan, M.C.; Lebecque, S.J.; Renno, T. TLR3 can directly trigger apoptosis in human cancer cells. J. Immunol. 2006, 176, 4894–4901.
- Gambara, G.; Desideri, M.; Stoppacciaro, A.; Padula, F.; De Cesaris, P.; Starace, D.; Tubaro, A.; Del Bufalo, D.; Filippini, A.; Ziparo, E.; et al. TLR3 engagement induces IRF-3-dependent apoptosis in androgen-sensitive prostate cancer cells and inhibits tumour growth in vivo. J. Cell. Mol. Med. 2015, 19, 327–339.
- Umansky, V.; Schirrmacher, V. Nitric oxide-induced apoptosis in tumor cells. Adv. Cancer Res. 2001, 82, 107–131.
- Yadav, J.; Dikshit, N.; Ismaeel, S.; Qadri, A. Innate Activation of IFN-γ-iNOS Axis During Infection with Salmonella Represses the Ability of T Cells to Produce IL-2. Front. Immunol. 2020, 11, 514.
More