Microtubule poisons, as is the case with other antitumor drugs, routinely promote autophagy in tumor cells. However, the nature and function of the autophagy, in terms of whether it is cytoprotective, cytotoxic or nonprotective, cannot be predicted; this likely depends on both the type of drug studied as well as the tumor cell under investigation. The microtubule poisons continue to play a central role in the clinical treatment of both solid tumors or hematologic malignancies [35]. However, tumor cells can develop resistance by a number of mechanisms such as altered microtubule binding and efflux via the multidrug resistance pump family of transporters [36,37].
- autophagy
- microtubule poison
- cytoprotective
- cytotoxic
1. Introduction
2. Direct Involvement of Microtubules in Autophagy
When microtubule-associated protein 1A/1B-light chain 3 (MAP1LC3, LC3) was identified as a key factor in the induction of autophagy, the Yoshimori laboratory had proposed the idea that microtubules might be involved in the progression of autophagy by affecting the efficient transport of autophagosomes to mammalian cells [38][12]. It was later confirmed that microtubules participate in the fusion of autophagosomes with lysosomes and in the formation of late autophagosomes [39,40][13][14]. The importance of microtubules in autophagic flux has long been recognized, and their role in autophagy initiation, trafficking, and lysosomal fusion has been continuously revealed in the last two decades. In addition to LC3, other autophagy-related proteins such as ULK1, Beclin-1, WIP1, ATG5, and ATG12 that are involved in autophagosome formation were found to be associated with microtubules as well [41,42,43][15][16][17]. Dynamic changes in microtubule and post-translational modifications of microtubule play an important role in regulating starvation-induced autophagy, as microtubule acetylation occurs prior to autophagosome formation in response to nutrient deficiency. Acetylation modification signals kinesin recruitment to microtubules, followed by JNK activation and Beclin-1 release from the Beclin-1-Bcl-2 complex to initiate autophagy [41][15]. AMBRA1 is a key factor in the regulation of autophagy in vertebrates. AMBRA1 promotes the interaction of Beclin-1 with its target lipid kinase, VPS34, which mediates autophagosome nucleation [44][18]. Moreover, AMBRA1 serves as a direct regulatory link between ULK1 and Beclin-1-VPS34, which is required for the localization and activity of the intracellular core complex. Once the autophagosome is formed, it moves along the microtubule in both directions (minus-end and plus-end) via kinesin motor complexes, accumulating at the microtubule-organizing center, and eventually moving towards the lysosome [39,40,45][13][14][19]. Their centripetal movement is dependent on the motor protein dynein and is important for their fusion with lysosomes. In addition to the function of microtubules mediating autophagosome transport, there is a strict regulatory relationship between cytoskeletal dynamics and autophagosome formation [43][17]. The effect of microtubules on the fusion of autophagosomes with lysosomes is controversial. One view is that microtubule dynamics does not affect the fusion of autophagosomes with lysosomes and that this fusion can occur in the presence of microtubule poisons [46,47][20][21]. However, by combining real-time observation and microinjection techniques, other investigators proposed that after formation, autophagosomes utilize a dynein-microtubule system to rapidly move toward lysosomes located near the centrosome [40][14]. When cells are starved for glucose, the cyclin-dependent kinase inhibitor p27Kip1 (p27) promotes autophagy by maintaining elevated microtubule acetylation via an ATAT1-dependent mechanism to promote autophagosome transport along microtubule trails [48][22]. In addition to their classification into the two main categories, namely microtubule destabilizing agents and microtubule stabilizing agents, microtubule poisons are then subdivided into the following categories according to their specific binding domains to microtubule proteins (Figure 1): colchicine site, vinca alkaloid site, taxane site, laulimalide site and epothilone site. Analysis of autophagy induced by different microtubule poisons in tumor therapy is based on these subcategories.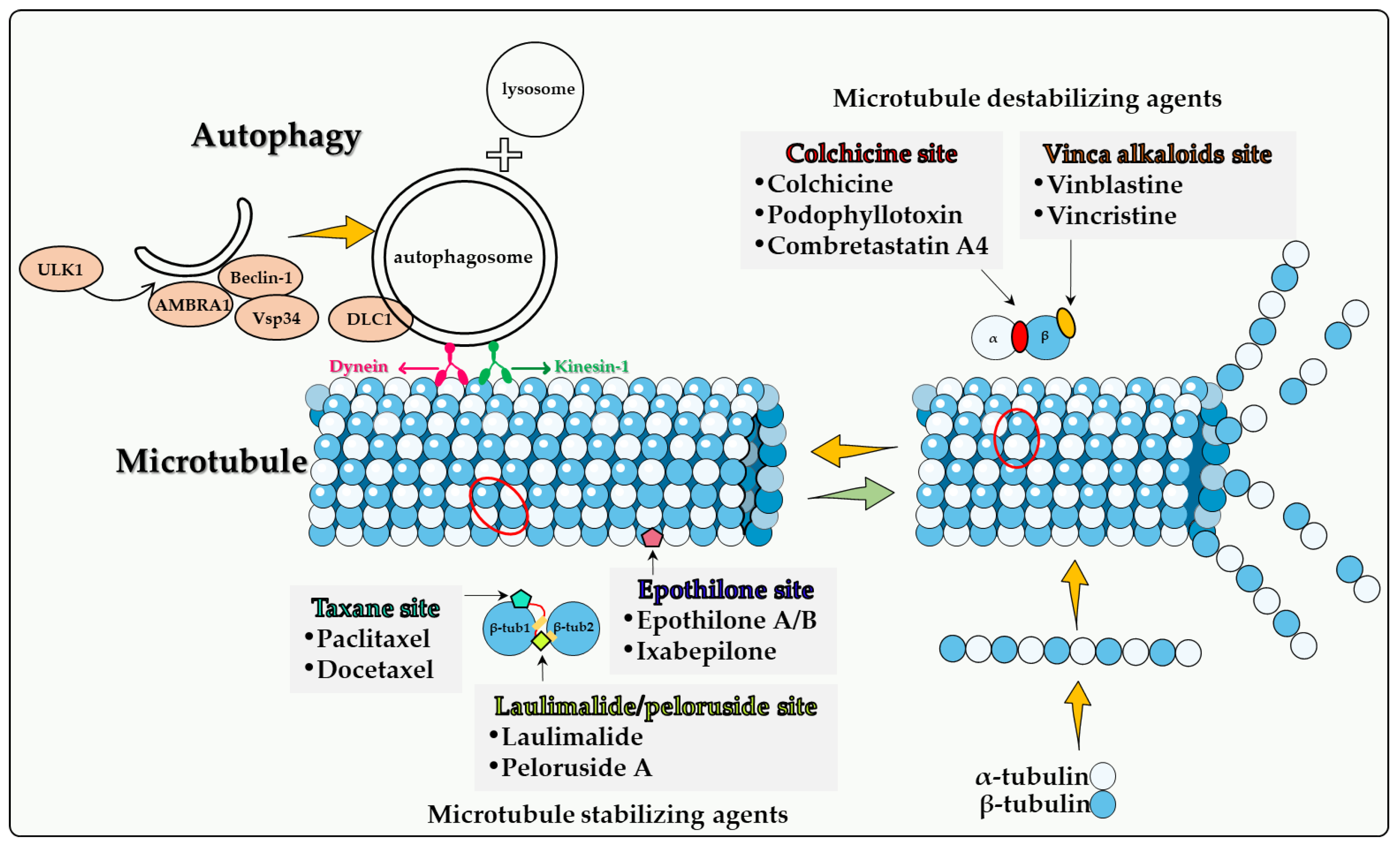
2.1. Colchicine Site
Colchicine. Colchicine, an alkaloid isolated from plants belonging to the genus Colchicum (Autumn crocus), is a classical antimitotic compounds, but is not actually used in cancer therapeutics due to its high degree of toxicity [49][23]. Colchicine blocks mitotic cells in metaphase through the formation of poorly reversible tubulin–colchicine complexes. In addition, colchicine binds to microtubule ends, thereby preventing elongation [49][23]. Although colchicine has potent antitumor properties that are attributed to the irreversible binding with tubulin causing cell cycle inhibition and the induction of apoptosis, colchicine lacks selectivity against tumor cells. This limitation has led to the development of colchicine derivatives as well as colchicinoid prodrugs with less toxicity and more specific targeting to tumor cells [51][24]. JG-03-14, a substituted pyrrole colchicine mimetic that binds to the colchicine site of tubulin, induced a significant reduction in the viability of MCF-7 and MDA-MB-231 cells [52][25]. Importantly, JG-03-14 was able to induce autophagy in up to 70% of the MCF-7 cell population by the third day of treatment with minimal apoptosis (as evidenced by the TUNEL assay). Conversely, JG-03-14 induced a significant amount of apoptosis by the third day of treatment in the MDA-MB231 cell line, but also demonstrated a significant degree of autophagy, based on acridine orange staining [52][25]. These findings suggest that JG-03-14 may have induced autophagic cell death in these breast tumor cell lines, although the precise function of the autophagy was not directly assessed. Combretastatin A-4. Combretastatins represent a large family of bioactive stilbenes, dihydrostilbenes, phenanthrenes and macrocyclic lactones named Combretastatins A, B, C, and D, respectively [63][26]. Combretastatins are isolated from Combretum caffrumtree and show favorable anticancer activities. A well-studied member of this drug family is Combretastatin A-4 (CA-4), a tubulin-depolymerizing agent that binds at the colchicine binding site on the β-tubulin subunit of tubulin, resulting in depolymerization and destabilization of tubulin polymers of the cytoskeleton, causing an increase in vasculature permeability and disruption of the tumor blood flow [63,64,65,66][26][27][28][29]. Combretastatin A4 phosphate is a prodrug for CA-4 that has shown a potential anti-cancer effect in Phase I clinical trials [70][30]. Hoang et al. [71][31] showed that the anti-tumor effect of CA-4 phosphate is enhanced in autophagy-defective PC3 prostate cancer xenografts (developed with retrovirally transducing PC-3 cells with ATG4BC74A, an inactive and dominant-negative mutant of the autophagy related gene atg4B) compared with controls. Significant central necrosis as well as a higher number of senescent cells were evident in autophagy-defective PC3 xenografts both 24 h and 1 week following CA4P treatment, indicating the possible role of autophagy inhibition (i.e., cytoprotective autophagy) in enhancing the antitumor effects of CA4 phosphate via lowering the threshold of peripheral tumor cells to tolerate the CA4 phosphate-induced metabolic stress [71,72][31][32]. Taken together, these findings suggest that combretastatin-mediated autophagy is largely cytoprotective but can also be dependent on both the cancer type as well as the specific cell line studied.2.2. Vinca Alkaloid Site
Vinca alkaloids are a class of organic compounds that were isolated from the leaves of the Madagascar periwinkle plant, Catharanthus roseus. Five distinct vinca compounds with significant antineoplastic activities have been identified, specifically vinblastine, vincristine, vindesine, vinorelbine, and vinflunine. Vinblastine and vincristine continue to be two of the most commonly used anticancer agents. These drugs are structurally related except that vincristine contains an aldehydic functional group attached to the nitrogen of the indole moiety whereas vinblastine contains a methyl group. This minor difference in structure results in significant differences in both the antineoplastic activities and the toxicity between the two agents [73][33]. Vinblastine has been utilized in the clinical treatment of leukemia, non-Hodgkin’s and Hodgkin’s disease, breast cancer, testicular carcinoma, and small-cell lung cancer. Vincristine have been used for many years in the treatment of malignancies including acute lymphoblastic leukemia, B-cell lymphoma, metastatic melanoma, and Wilms’ tumor [74][34]. Vinca alkaloids bind the vinca binding site on microtubules causing microtubule interruption and dissociation [75][35]. Vinca alkaloids tend to demonstrate cell cycle specificity for the M-phase [76][36]. At low concentrations, these drugs decrease the rates of both growth and shortening at the microtubule assembly end, blocking mitosis, with the cells eventually undergoing apoptosis [77][37]. At high concentrations, vinca alkaloids cause microtubule depolymerization and the destruction of mitotic spindles. The dividing tumor cells show condensed chromosomes and appear to be blocked in mitosis [78][38]. Furthermore, vinca alkaloids exhibit antiangiogenic and antivascular activities, inducing potent vascular disruption, and ultimately leading to tumor necrosis [36,79][39][40]. The function of autophagy is likely to vary between different experimental tumor models. In some cases, vincristine cytotoxicity may be hindered by the cytoprotective form of autophagy, while in other cases cytotoxic autophagy is expressed.2.3. Taxane Site
Paclitaxel. Paclitaxel, an antimitotic agent originally extracted from the bark of the Pacific yew tree, was identified and developed by Dr. Susan Band Horwitz in 1979 [90][41]. Paclitaxel binds to the β-tubulin subunit and forms stable and nonfunctional microtubules, thereby blocking cancer cell growth by interrupting cell division at the metaphase/anaphase transition, resulting in cell death [91,92][42][43]. This is a mechanism quite different from that of vincristine and vinblastine, which cause the disassembly of microtubules. Since its approval by the FDA, Taxol, a semisynthetic form of paclitaxel, has expanded treatment options for patients with breast [93][44] and ovarian cancers [94][45]. Non-small cell lung cancer, pancreatic cancer, and AIDS-related Kaposi sarcoma are all sensitive to Taxol as well [95,96][46][47]. The autophagy caused by paclitaxel in different types of tumor cells appears to be drastically different. Zou et al. found that paclitaxel cannot induce autophagy in SKBr3 and MDA-MB-231 breast cancer cells, unless ARHI (DIRAS3) is re-expressed in the cells [97][48]. Veldhoen et al. found that low concentrations of paclitaxel inhibited autophagy. It was suggested that paclitaxel-induced mitotic arrest leads to decreased autophagic flux through phosphorylation and inhibition of Vps34 and subsequently results in aberrant autophagosome trafficking and localization, which in turn inhibits autophagosome degradation. By detecting autophagosome formation of GFP-LC3 fluorescence in single cells and cell death using flow cytometry, these investigators demonstrated that 3-MA, siRNA ATG7, or siRNA VPS34 reduced paclitaxel-induced apoptosis, suggesting that the blocked autophagy still plays a key role in paclitaxel-induced cell death [98][49]. These studies appear to support a direct role of (cytotoxic) autophagy in paclitaxel-induced cell death.2.4. Epothilone Site
Epothilone A and epothilone B. Epothilones are a type of natural cytotoxic compound belonging to 16-member natural macrolides. Thus far, six types of epothilone and derivatives have been reported. Epothilone A and B were first isolated from myxobacterium Sorangium cellulosum, and the efficiency of epothilone B was shown to be higher than that of epothilone A [107][50]. Similar to paclitaxel, epothilone also exerts its antitumor activity by stabilizing cell microtubules, but the binding sites of epothilone and paclitaxel are different. Among the five oxygen-containing polar groups constituting the macrocycle of epothilone, only C7-OH is located near the similar C7-OH part of paclitaxel, and the polymerization activity of epothilone B is 2 to 10 times higher than that of paclitaxel [108][51]. In addition, epothilone is less susceptible to multidrug resistance pump-mediated efflux compared to paclitaxel, and the expression of MDR is not altered in epothilone-resistant cell lines, implying a wider choice for chemo-resistant patients [109,110][52][53].2.5. Laulimalide/Peloruside Site
Laulimalide and Peloruside A. Laulimalide is a potent microtubule stabilizer that was originally isolated from the sponge Cacospongia mycofijiensis [117][54]. Peloruside A was isolated from the New Zealand marine sponge Mycale hentscheli in 2000 [118][55]. Researchers found that peloruside A and laulimalide compete for the same or overlapping binding sites, not taxane site but the M-loop of β1 and loop H1−B2 of β2 [119,120,121][56][57][58]. In contrast to paclitaxel, peloruside A and laulimalide are not substrates for the multidrug resistance P-glycoprotein efflux pump and are not affected of β-tubulin mutations in the taxane binding site [119][56]. These properties indicate that laulimalide and peloruside A have the potential to treat paclitaxel resistant tumors, but no clinical trials are currently in progress due to a limited drug supply, unstable drug structure [122][59] and the lack of antitumor activity studies in vivo.3. Summary
Although many new cancer treatments have been developed in recent years, such as targeted therapy and immunotherapy, microtubule poisons remain a critical class of first-line chemotherapeutic agents. However, the relationship(s) between microtubule poisons and autophagy are exceedingly complex. This is likely due to a number of factors including that (a) microtubules play a direct role in the autophagic process; (b) the different microtubule poisons do not all act at the identical microtubule binding sites, as indicated in Figure 1; (c) autophagy cannot have only one (cytoprotective) function, but (at least) three others, termed cytotoxic, cytostatic and nonprotective. Although the cytoprotective function is often clearly induced, both the cytotoxic and nonprotective function have also been identified in response to these agents. This may be dependent upon the chemical structure of the drug as well as the experimental cell line being utilized. final and critical issue is that the literature evaluating the role of autophagy in response to microtubule poisons in tumor cells has generally relied on pharmacologic autophagy inhibitors such as CQ (or HCQ) and 3-MA drugs, which are not exclusively autophagy inhibitors [106][60]. In the absence of studies utilizing genetic autophagy inhibition, inferences related to the nature of the autophagy are not sufficiently well supported to be conclusive.
References
- Caplow, M. Microtubule dynamics. Curr. Opin. Cell Biol. 1992, 4, 58–65.
- Carlier, M.F.; Hill, T.L.; Chen, Y. Interference of GTP hydrolysis in the mechanism of microtubule assembly: An experimental study. Proc. Natl. Acad. Sci. USA 1984, 81, 771–775.
- Jánosi, I.M.; Chrétien, D.; Flyvbjerg, H. Structural microtubule cap: Stability, catastrophe, rescue, and third state. Biophys. J. 2002, 83, 1317–1330.
- Mitchison, T.J.; Kirschner, M.W. Dynamic instability of microtubule growth. Nature 1984, 312, 237–242.
- McKean, P.G.; Vaughan, S.; Gull, K. The extended tubulin superfamily. J. Cell. Sci. 2001, 114, 2723–2733.
- Goodson, H.V.; Jonasson, E.M. Microtubules and microtubule-associated proteins. Cold Spring Harb. Perspect. Biol. 2018, 10, a022608.
- Cutts, J.H.; Beer, C.T.; Noble, R.L. Biological properties of Vincaleukoblastine, an alkaloid in Vinca rosea Linn, with reference to its antitumor action. Cancer Res. 1960, 20, 1023–1031.
- Schneider, F.; Pan, L.; Ottenbruch, M.; List, T.; Gaich, T. The chemistry of nonclassical taxane diterpene. Acc. Chem. Res. 2021, 54, 2347–2360.
- Yang, C.H.; Horwitz, S.B. Taxol(R): The first microtubule stabilizing agent. Int. J. Mol. Sci. 2017, 18, 1733.
- Huitorel, P. From cilia and flagella to intracellular motility and back again: A review of a few aspects of microtubule-based motility. Biol. Cell 1988, 63, 249–258.
- Forth, S.; Kapoor, T.M. The mechanics of microtubule networks in cell division. J. Cell Biol. 2017, 216, 1525–1531.
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728.
- Jahreiss, L.; Menzies, F.M.; Rubinsztein, D.C. The itinerary of autophagosomes: From peripheral formation to kiss-and-run fusion with lysosomes. Traffic 2008, 9, 574–587.
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent Movement of Autophagosomes Mediates Efficient Encounters with Lysosomes. Cell Struct. Funct. 2008, 33, 109–122.
- Geeraert, C.; Ratier, A.; Pfisterer, S.G.; Perdiz, D.; Cantaloube, I.; Rouault, A.; Pattingre, S.; Proikas-Cezanne, T.; Codogno, P.; Poüs, C. Starvation-induced hyperacetylation of tubulin is required for the stimulation of autophagy by nutrient deprivation. J. Biol. Chem. 2010, 285, 24184–24194.
- Luo, S.; Garcia-Arencibia, M.; Zhao, R.; Puri, C.; Toh, P.P.; Sadiq, O.; Rubinsztein, D.C. Bim Inhibits autophagy by recruiting beclin 1 to microtubules. Mol. Cell 2012, 47, 359–370.
- Di Bartolomeo, S.; Corazzari, M.; Nazio, F.; Oliverio, S.; Lisi, G.; Antonioli, M.; Pagliarini, V.; Matteoni, S.; Fuoco, C.; Giunra, L. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J. Cell Biol. 2010, 191, 155–168.
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125.
- Kast, D.J.; Dominguez, R. The cytoskeleton–autophagy connection. Curr. Biol. 2017, 27, R318–R326.
- Fass, E.; Shvets, E.; Degani, I.; Hirschberg, K.; Elazar, Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J. Biol. Chem. 2006, 281, 36303–36316.
- Köchl, R.; Hu, X.W.; Chan, E.Y.W.; Tooze, S.A. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic 2006, 7, 129–145.
- Nowosad, A.; Creff, J.; Jeannot, P.; Culerrier, R.; Codogno, P.; Manenti, S.; Nguyen, L.; Besson, A. p27 controls autophagic vesicle trafficking in glucose-deprived cells via the regulation of ATAT1-mediated microtubule acetylation. Cell Death Dis. 2021, 12, 481.
- Bhattacharyya, B.; Panda, D.; Gupta, S.; Banerjee, M. Anti-mitotic activity of colchicine and the structural basis for its interaction with tubulin. Med. Res. Rev. 2008, 28, 155–183.
- Sivakumar, G. Colchicine semisynthetics: Chemotherapeutics for cancer? Curr. Med. Chem. 2013, 20, 892–898.
- Arthur, C.R.; Gupton, J.T.; Kellogg, G.; Yeudall, W.A.; Cabot, M.C.; Newsham, I.F.; Gewirtz, D.A. Autophagic cell death, polyploidy and senescence induced in breast tumor cells by the substituted pyrrole JG-03-14, a novel microtubule poison. Biochem. Pharmacol. 2007, 74, 981–991.
- Karatoprak, G.Ş.; Küpeli Akkol, E.; Genç, Y.; Bardakcı, H.; Yücel, Ç.; Sobarzo-Sánchez, E. Combretastatins: An overview of structure, probable mechanisms of action and potential applications. Molecules 2020, 25, 2560.
- Dowlati, A.; Robertson, K.; Cooney, M.; Petros, W.P.; Stratford, M.; Jesberger, J.; Rafie, N.; Overmoyer, B.; Makkar, V.; Stambler, B.; et al. A phase I pharmacokinetic and translational study of the novel vascular targeting agent combretastatin a-4 phosphate on a single-dose intravenous schedule in patients with advanced cancer. Cancer Res. 2002, 62, 3408–3416.
- Simoni, D.; Romagnoli, R.; Baruchello, R.; Rondanin, R.; Rizzi, M.; Pavani, M.G.; Alloatti, D.; Giannini, G.; Marcellini, M.; Riccioni, T.; et al. Novel combretastatin analogues endowed with antitumor activity. J. Med. Chem. 2006, 49, 3143–3152.
- West, C.M.; Price, P. Combretastatin A4 phosphate. Anticancer. Drugs 2004, 15, 179–187.
- Rustin, G.J.; Galbraith, S.M.; Anderson, H.; Stratford, M.; Folkes, L.K.; Sena, L.; Gumbrell, L.; Price, P.M. Phase I clinical trial of weekly combretastatin A4 phosphate: Clinical and pharmacokinetic results. J. Clin. Oncol. 2003, 21, 2815–2822.
- Hoang, V.C.; Chow, A.; Emmenegger, U. Abstract 1688: Autophagy inhibition enhances the antitumor effects of combretastatin A4 phosphate (CA4P). Cancer Res. 2013, 73 (Suppl. S8), 1688.
- Greene, L.M.; Meegan, M.J.; Zisterer, D. Combretastatins: More Than Just Vascular Targeting Agents? J. Pharmacol. Exp. Ther. 2015, 355, 212–227.
- Agrawal, K. Vinblastine. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–4.
- Lee, C.-T.; Huang, Y.-W.; Yang, C.-H.; Huang, K.-S. Drug delivery systems and combination therapy by using vinca alkaloids. Curr. Top. Med. Chem. 2015, 15, 1491–1500.
- Downing, K.H. Structural Basis for the Interaction of Tubulin with Proteins and Drugs that Affect Microtubule Dynamics. Annu. Rev. Cell Dev. Biol. 2000, 16, 89–111.
- Silverman, J.A.; Deitcher, S.R. Marqibo® (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother. Pharmacol. 2013, 71, 555–564.
- Moudi, M.; Go, R.; Yien, C.Y.S.; Nazre, M. Vinca alkaloids. Int. J. Prev. Med. 2013, 4, 1231–1235.
- Silvestri, R. New prospects for vinblastine analogues as anticancer agents. J. Med. Chem. 2013, 56, 625–627.
- Krause, W. Resistance to anti-tubulin agents: From vinca alkaloids to epothilones. Cancer Drug Resist. 2019, 2, 2–106.
- Carlson, R.O. New tubulin targeting agents currently in clinical development. Expert Opin. Investig. Drugs 2008, 17, 707–722.
- Schiff, P.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667.
- Horwitz, S.B. Mechanism of action of taxol. Trends Pharmacol. Sci. 1992, 13, 134–136.
- A Jordan, M.; Toso, R.J.; Thrower, D.; Wilson, L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc. Natl. Acad. Sci. 1993, 90, 9552–9556.
- Raveendran, R.S.; Baby, S. Resistance to intervention: Paclitaxel in breast cancer. Mini-Rev. Med. Chem. 2021, 21, 1237–1268.
- Baird, R.D.; Tan, D.S.P.; Kaye, S.B. Weekly paclitaxel in the treatment of recurrent ovarian cancer. Nat. Rev. Clin. Oncol. 2010, 7, 575–582.
- Chen, Q.; Xu, S.; Liu, S.; Wang, Y.; Liu, G. Emerging nanomedicines of paclitaxel for cancer treatment. J. Control. Release 2022, 342, 280–294.
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. J. Am. Med. Assoc. 2021, 326, 851–862.
- Zou, C.-F.; Jia, L.; Jin, H.; Yao, M.; Zhao, N.; Huan, J.; Lu, Z.; Bast, R.C.; Feng, Y.; Yu, Y. Re-expression of ARHI (DIRAS3) induces autophagy in breast cancer cells and enhances the inhibitory effect of paclitaxel. BMC Cancer 2011, 11, 22.
- Veldhoen, R.A.; Banman, S.L.; Hemmerling, D.R.; Odsen, R.; Simmen, T.; Simmonds, A.J.; Underhill, D.A.; Goping, I.S. The chemotherapeutic agent paclitaxel inhibits autophagy through two distinct mechanisms that regulate apoptosis. Oncogene 2013, 32, 736–746.
- Gerth, K.; Bedorf, N.; Höfle, G.; Irschik, H.; Reichenbach, H. Epothilons A and B: Antifungal and cytotoxic compounds from Sorangium cellulosum (Myxobacteria). Production, physico-chemical and biological properties. J. Antibiot. 1996, 49, 560–563.
- Nettles, J.H.; Downing, K.H. The binding mode of epothilone A on alpha, beta-tubulin by electron crystallography. Science 2004, 305, 866–869.
- Chou, T.-C.; Zhang, X.-G.; Harris, C.R.; Kuduk, S.D.; Balog, A.; Savin, K.A.; Bertino, J.R.; Danishefsky, S.J. Desoxyepothilone B is curative against human tumor xenografts that are refractory to paclitaxel. Proc. Natl. Acad. Sci. USA 1998, 95, 15798–15802.
- Fumoleau, P.; Coudert, B.; Isambert, N.; Ferrant, E. Novel tubulin-targeting agents: Anticancer activity and pharmacologic profile of epothilones and related analogues. Ann. Oncol. 2007, 18 (Suppl. S5), v9–v15.
- Mooberry, S.L.; Tien, G.; Hernandez, A.H.; Plubrukarn, A.; Davidson, B.S. Laulimalide and isolaulimalide, new paclitaxel-like microtubule-stabilizing agents. Cancer Res. 1999, 59, 653–660.
- West, L.M.; Northcote, P.T.; Battershill, C.N. Peloruside A: A Potent Cytotoxic Macrolide Isolated from the New Zealand Marine Sponge Mycale sp. J. Org. Chem. 2000, 65, 445–449.
- Gaitanos, T.N.; Buey, R.M.; Díaz, J.F.; Northcote, P.T.; Teesdale-Spittle, P.; Andreu, J.M.; Miller, J.H. Peloruside A does not bind to the taxoid site on beta-tubulin and retains its activity in multidrug-resistant cell lines. Cancer Res. 2004, 64, 5063–5067.
- Prota, A.E.; Bargsten, K.; Northcote, P.T.; Marsh, M.; Altmann, K.H.; Miller, J.H.; Díaz, J.F.; Steinmetz, M.O. Structural basis of microtubule stabilization by laulimalide and peloruside A. Angew. Chem. Int. Ed. Engl. 2014, 53, 1621–1625.
- Castro-Alvarez, A.; Pineda, O.; Vilarrasa, J. Further insight into the interactions of the cytotoxic macrolides laulimalide and peloruside a with their common binding site. ACS Omega 2018, 3, 1770–1782.
- Gollner, A.; Altmann, K.-H.; Gertsch, J.; Mulzer, J. The laulimalide family: Total synthesis and biological evaluation of neolaulimalide, isolaulimalide, laulimalide and a nonnatural analogue. Chem. A Eur. J. 2009, 15, 5979–5997.
- Pickard, R.D.; Spencer, B.H.; McFarland, A.J.; Bernaitis, N.; Davey, A.K.; Perkins, A.V.; Chess-Williams, R.; McDermott, C.M.; Forbes, A.; Christie, D.; et al. Paradoxical effects of the autophagy inhibitor 3-methyladenine on docetaxel-induced toxicity in PC-3 and LNCaP prostate cancer cells. Naunyn-Schmiedebergs Arch. Fur Exp. Pathol. Und Pharmakol. 2015, 388, 793–799.