Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Rita Xu and Version 2 by Rita Xu.
Fibrosis results from defective wound healing processes often seen after chronic injury and/or inflammation in a range of organs. Progressive fibrotic events may lead to permanent organ damage/failure. The hallmark of fibrosis is the excessive accumulation of extracellular matrix (ECM), mostly produced by pathological myofibroblasts and myofibroblast-like cells. The Hippo signaling pathway is an evolutionarily conserved kinase cascade, which has been described well for its crucial role in cell proliferation, apoptosis, cell fate decisions, and stem cell self-renewal during development, homeostasis, and tissue regeneration.
- tissue fibrosis
- fibroblast
1. Introduction
Impaired tissue repair processes due to the dysregulation of molecular and cellular events after organ injury lead to organ fibrosis, a common cause of organ failure after chronic injury and/or inflammation [1]. A hallmark of fibrosis is the excessive deposition of the collagen-rich extracellular matrix (ECM), especially due to an imbalance between collagen synthesis and remodeling. One of the key processes in fibrosis is the differentiation/transition of fibroblasts into myofibroblasts, the main fibrotic cellular phenotype, which produces a series of ECM components such as collagens, laminins, and fibronectins [2][3]. Numerous cell types, including epithelial and endothelial cells are also able to transdifferentiate into myofibroblastic phenotype (such as epithelial-mesenchymal transition and endothelial-mesenchymal transition) during organ fibrogenesis [4]. Recent studies have implicated the contribution of Hippo signaling pathway components in the fibrosis of various tissues, including the lung [5][6][7], liver [8][9][10][11][12][13][14], kidney [15][16][17], and heart [18][19][20]. The Hippo signaling pathway was first identified and illustrated in Drosophila. In mammals, the key components of the pathway are highly conserved, including several serine/threonine kinases and transcriptional factors, which are orthologs of Drosophila proteins. The transcriptional regulator Yes-associated protein (YAP) and its co-activator PDZ-binding motif (TAZ/WWTR1) are the main effectors of this pathway. In response to physiological or pathological stimuli, sterile 20-like protein kinase (MST1/2) forms complexes with the adaptor protein Salvador 1 (SAV1) that phosphorylates large tumor suppressors (LATS1/2) and LATS1/2-interacting protein MOB kinase activator 1 (MOB1). The phosphorylated LATS1/2–MOB1 complexes, in turn, phosphorylate YAP and TAZ, resulting in the cytoplasmic retention or polyubiquitination and subsequent degradation of YAP/TAZ by proteasomes during autophagy (Figure 1). In contrast, dephosphorylation of upstream kinase cascade drives the nuclear trafficking of YAP and TAZ, where they can interact with numerous transcription factors, including TEA domain DNA-binding family members (TEAD1–4), and regulate the expression of Hippo pathway target genes responsible for cell proliferation, survival, and differentiation [21][22][23].
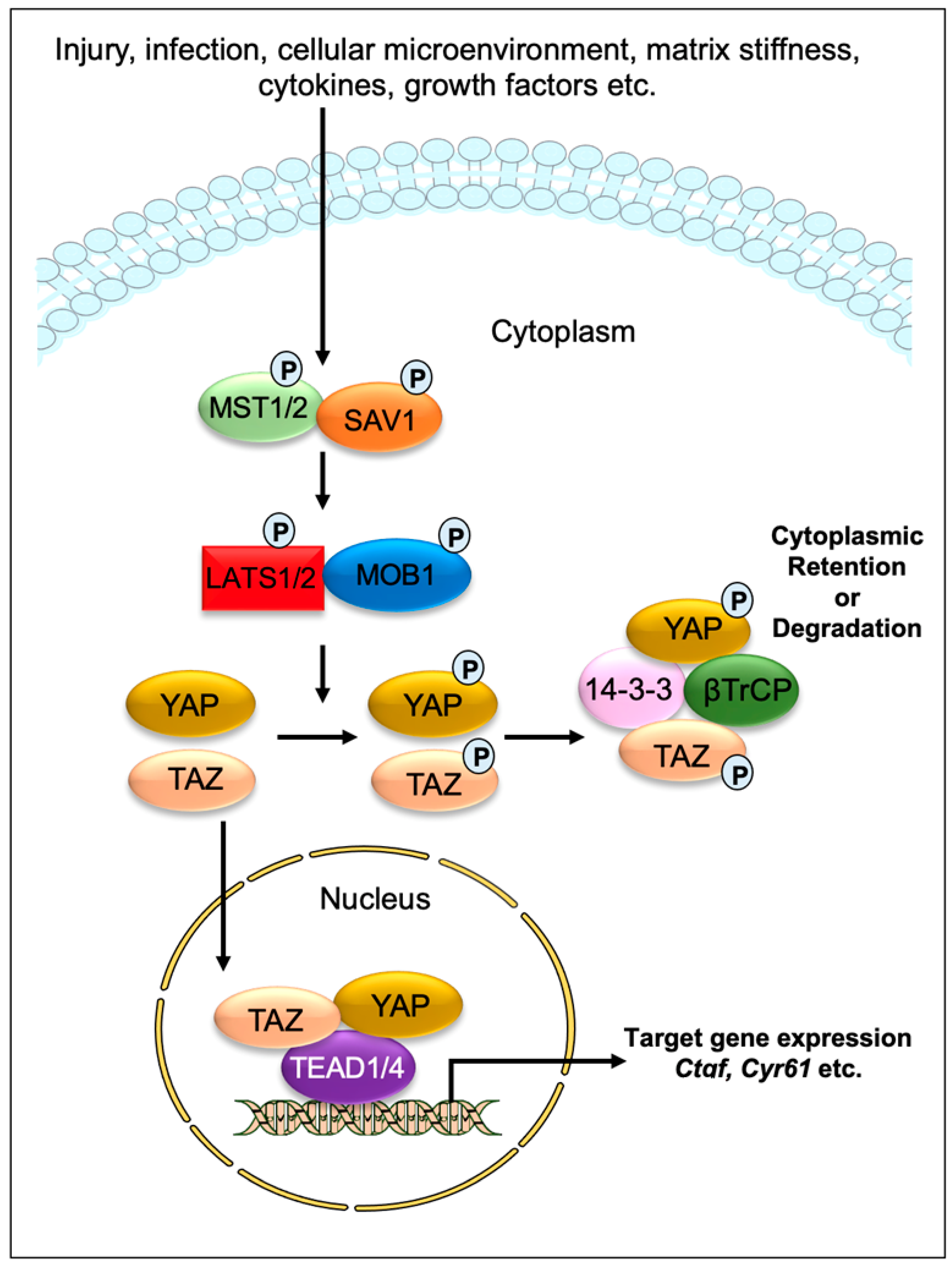
Figure 1. Graphical demonstration of the Hippo signaling pathway. Various physiological and pathological signals can induce the Hippo signaling pathway. In mammals, the core elements of the Hippo pathway mainly consist of serine/threonine kinases, transcriptional factors, and their cofactors. The transcriptional factors Yes-associated protein (YAP) and its coactivator PDZ-binding motif (TAZ/WWTR1) are the key effectors of the Hippo pathway. After physiological or pathological stimuli, sterile 20-like protein kinase (MST1/2) interacts, phosphorylates, and forms complexes with the adaptor protein Salvador 1 (SAV1), which phosphorylates large tumor suppressor (LATS1/2) and LATS1/2-interacting protein MOB kinase activator 1 (MOB1). The phosphorylated LATS1/2–MOB1 complex then phosphorylates YAP and TAZ, which promotes cytoplasmic retention or polyubiquitination and consequent degradation of YAP/TAZ by proteasomes during autophagy. However, dephosphorylation of upstream kinases leads to the nuclear translocation of YAP and TAZ, where they can interact with various transcription factors including TEA domain DNA-binding family members (TEAD1–4) and regulate the expression of Hippo pathway target genes such as Ctgf and Cyr61.
The Hippo signaling pathway has been extensively studied for its role as an organ size controller that regulates cell proliferation, apoptosis, cell fate decisions, and stem cell self-renewal during development, homeostasis, regeneration, and cancer formation in numerous mammalian organs [21][22][24][25][26]. A growing number of studies have reported that the cascade components also play a critical role in fibrotic diseases. For example, the nuclear YAP/TAZ in resident cardiac fibroblasts obtained from preclinical myocardial infarction (MI) models [18][19][27], are sufficient to direct the transdifferentiation of cardiac fibroblasts into pathological myofibroblasts [27][28]. Similarly, YAP/TAZ are important regulators of pathological fibroblast activation in pulmonary fibrosis [5] and hepatic stellate cell activation in liver fibrosis [11][12][13][14]. Furthermore, activated YAP/TAZ in interstitial myofibroblasts promotes kidney fibrosis [16], while hyperactivated YAP/TAZ in non-fibroblast cells, such as macrophages [29][30], epithelial cells [31][32][33], and hepatocytes [8][10] contribute to the pathogenesis of fibrosis. Consistent with these findings showing the fibrotic effects of YAP/TAZ, their inactivation was found to be beneficial for preventing myofibroblast formation and fibrosis development [34]. Like YAP/TAZ, Hippo kinases are also known to contribute to a range of fibrotic diseases [20][35][36][37][38]. As such, treatment of mice with an MST1/2 blocker XMU-MP-1 inhibits pressure-induced cardiac hypertrophy and fibrosis (Table 1) [39]. Similarly, LATS1/2-mutant cardiac fibroblasts are less prone to develop into fibrotic phenotype after infarction-induced injury [35], while deletion of LATS1/2 stimulates YAP/TAZ activation in myofibroblasts and aggravates kidney fibrosis [40]. Similarly, genetic inactivation of SAV1 in renal tubule cells promotes renal interstitial fibrosis [36]. In fibrotic diseases, various profibrotic signals can induce the function of the Hippo effectors. For example, stiff substrates drive the nuclear localization of YAP and TAZ in fibroblasts and regulates their differentiation into myofibroblasts that promote profibrotic ECM synthesis; on the other hand, soft substrates prevent ECM synthesis. [5][15][17][41].
2. Hippo Signaling Pathway in Cardiac Fibrosis
The fibrotic response is a crucial contributor to heart failure (HF), which occurs in many types of cardiac diseases, including cardiac ischemia, myocardial infarction (MI), and cardiac hypertrophy. Understanding the mechanisms underlying fibrotic cardiac remodeling after injury remains a critical barrier to developing effective treatments for HF patients. An increasing number of studies have revealed important roles for Hippo signaling components in the development and progression of cardiac fibrosis [18][19][20][23][27][28][29][35][37][39][42][43][44][45][46][47][48]. While YAP/TAZ activation is crucial for driving cardiac fibrosis, their suppression was beneficial for preventing angiotensin II (AngII) or MI-induced fibrosis [18][19][29][44]. Cardiac fibroblasts are believed to be the major source of pathological ECM synthesis during cardiac remodeling. Elevated YAP and downregulated upstream Hippo kinase LATS1 were determined in the left ventricular tissue of HF patients, which was associated with cardiac fibroblast proliferation [42]. Recent studies also identified the activation of YAP and TAZ in resident cardiac fibroblasts from preclinical MI models (Figure 2) [18][19][27]. YAP and TAZ can directly induce the differentiation of fibroblasts into pathologic myofibroblasts [27][28]. Fibroblast-specific deletion of Yap/Taz using Col1a2Cre(ER)T mice [18] or deletion of only Yap using Tcf21MCM;YapF/F mice [19] showed reduced fibrotic and inflammatory responses in infarcted hearts. Similarly, Yap/Taz deletion resulted in an impaired profibrotic response in the fibroblasts from the infarcted heart. Fibroblast-specific Yap/Taz deficiency resulted in improved cardiac function in mice post-MI [18]. Loss of YAP also reduced myocardial fibrosis and cardiac dysfunction in response to chronic neuroendocrine stimulation by AngII. In vitro studies revealed that blocking of YAP/TAZ with siRNA or verteporfin abrogated TGF-β1-induced transition of fibroblasts into myofibroblasts and ECM (such as collagen and fibronectin) production [18]. In contrast, fibroblast-specific YAP overexpression using YAP5SA mice promoted inflammatory response, fibrosis, and hypertrophy in the infarcted heart [18]. Consistently, Francisco J et al. [28] described that targeted overexpression of YAP in cardiac fibroblasts with adeno-associated virus construct AAV-hTCF21-FLAG-Yap(S127A)-augmented myocardial inflammation and fibrosis in mice, resulting in declined cardiac function. Mechanistically, ras homolog gene family member A (RhoA) regulates AngII-induced YAP activation, which mediates the transition of cardiac fibroblasts into fibrotic myofibroblasts [19]. Moreover, YAP interacts with MRTF-A (myocardin-related transcription factor A) to facilitate the formation of α-smooth muscle actin (α-SMA)-positive myofibroblasts and profibrotic gene expression [19]. YAP/TAZ also regulate interleukin-33 (IL-33) to promote cardiac myofibroblasts formation [18]. These studies suggest that YAP and TAZ are crucially involved in regulating the formation of cardiac myofibroblasts and fibrosis.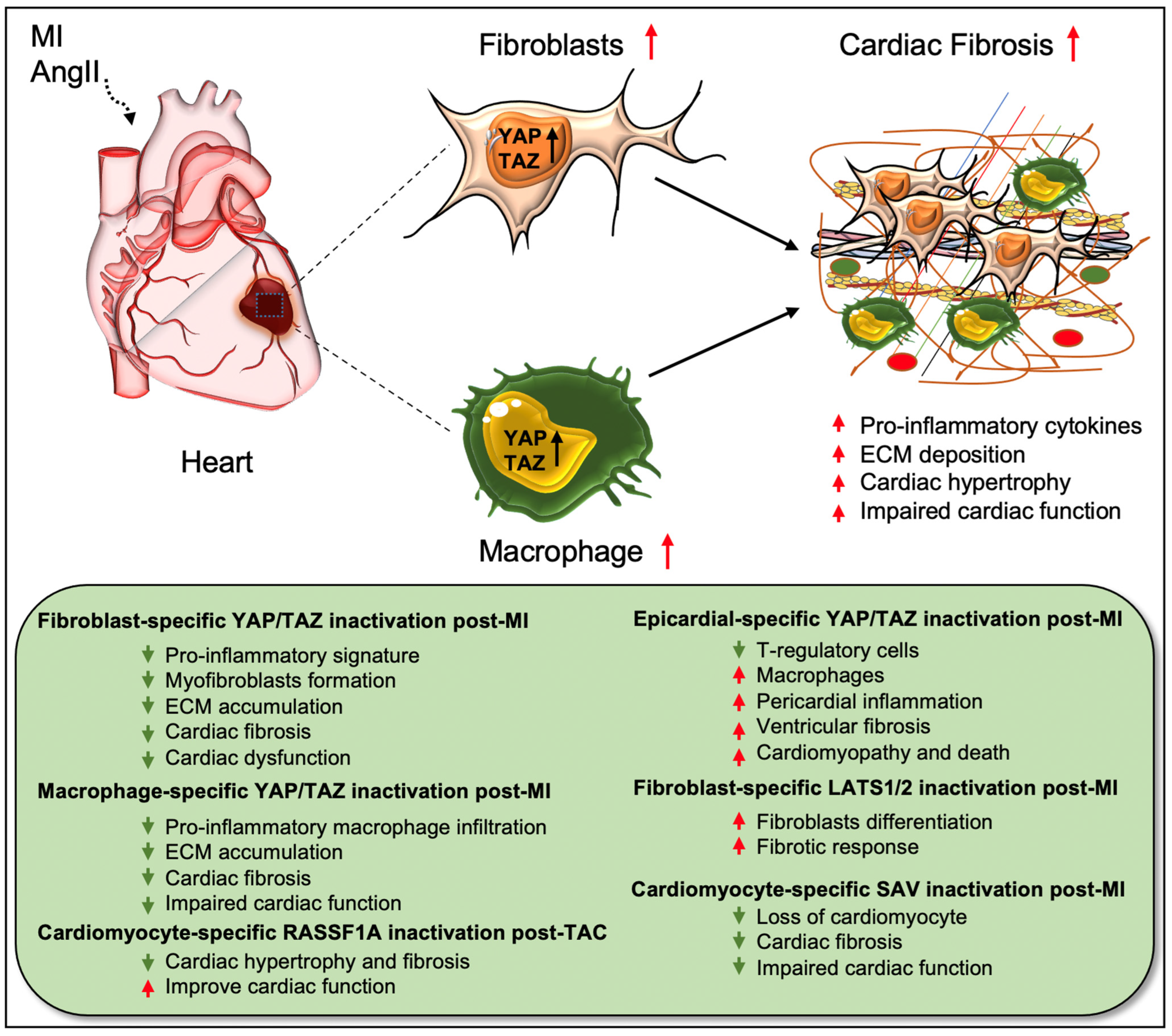
Figure 2. Role of Hippo/YAP pathway in cardiac fibrosis and repair. Activation of YAP/TAZ in fibroblasts and macrophages plays a crucial role in driving cardiac fibrosis after MI or AngII-induced injury. Fibroblast-specific loss of Yap/Taz or loss of only Yap improves cardiac outcome and reduces fibrotic response in infarcted hearts post-injury. Genetic ablation of Yap/Taz in fibroblasts blocks the proliferation of fibroblasts and their differentiation into pathologic myofibroblasts and disrupts macrophage-mediated inflammatory signature. In contrast, fibroblast-specific inactivation of upstream Hippo kinases Lats1/2 aggravates fibrotic response post-MI. Like fibroblasts, conditional ablation of Yap/Taz in macrophages leads to impairment of macrophage-mediated inflammatory response and thereby reduces cardiac fibrosis, resulting in improved cardiac function and hypertrophy post-MI. Consistently, cardiomyocyte-specific inactivation of Hippo kinase Sav reduces cardiomyocyte apoptosis and cardiac fibrosis, which causes improved cardiac function post-MI. Likewise, cardiomyocyte-specific deletion of Rassf1A reduces TAC-induced cardiac hypertrophy and fibrosis with improved cardiac function. Epicardium-specific deficiency of Yap/Taz reduces the infiltration of T-regulatory cells (a subclass of adaptive immune cells) while increasing the recruitment of macrophages (a subclass of innate immune cells) in the myocardium, which promotes pericardial inflammation and ventricular fibrosis after MI-injury that subsequently leads to cardiomyopathy and death.
References
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A common pathway to organ injury and failure. N. Engl. J. Med. 2015, 372, 1138–1149.
- Gabbiani, G. The myofibroblast: A key cell for wound healing and fibrocontractive diseases. Prog. Clin. Biol. Res. 1981, 54, 183–194.
- Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 2003, 200, 500–503.
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in Fibrosis: TGF-beta, WNT, and YAP/TAZ Converge. Front Med. 2015, 2, 59.
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinkovic, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L344–L357.
- Link, P.A.; Choi, K.M.; Diaz Espinosa, A.M.; Jones, D.L.; Gao, A.Y.; Haak, A.J.; Tschumperlin, D.J. Combined control of the fibroblast contractile program by YAP and TAZ. Am. J. Physiol. Lung Cell. Mol. Physiol. 2022, 322, L23–L32.
- Haak, A.J.; Kostallari, E.; Sicard, D.; Ligresti, G.; Choi, K.M.; Caporarello, N.; Jones, D.L.; Tan, Q.; Meridew, J.; Diaz Espinosa, A.M.; et al. Selective YAP/TAZ inhibition in fibroblasts via dopamine receptor D1 agonism reverses fibrosis. Sci. Transl. Med. 2019, 11, eaau6296.
- Mooring, M.; Fowl, B.H.; Lum, S.Z.C.; Liu, Y.; Yao, K.; Softic, S.; Kirchner, R.; Bernstein, A.; Singhi, A.D.; Jay, D.G.; et al. Hepatocyte Stress Increases Expression of Yes-Associated Protein and Transcriptional Coactivator With PDZ-Binding Motif in Hepatocytes to Promote Parenchymal Inflammation and Fibrosis. Hepatology 2020, 71, 1813–1830.
- Wang, X.; Cai, B.; Yang, X.; Sonubi, O.O.; Zheng, Z.; Ramakrishnan, R.; Shi, H.; Valenti, L.; Pajvani, U.B.; Sandhu, J.; et al. Cholesterol Stabilizes TAZ in Hepatocytes to Promote Experimental Non-alcoholic Steatohepatitis. Cell Metab. 2020, 31, 969–986.e967.
- Wang, X.; Zheng, Z.; Caviglia, J.M.; Corey, K.E.; Herfel, T.M.; Cai, B.; Masia, R.; Chung, R.T.; Lefkowitch, J.H.; Schwabe, R.F.; et al. Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2016, 24, 848–862.
- Li, C.; Zhang, R.; Zhan, Y.; Zheng, J. Resveratrol Inhibits Hepatic Stellate Cell Activation via the Hippo Pathway. Mediat. Inflamm. 2021, 2021, 3399357.
- Du, K.; Hyun, J.; Premont, R.T.; Choi, S.S.; Michelotti, G.A.; Swiderska-Syn, M.; Dalton, G.D.; Thelen, E.; Rizi, B.S.; Jung, Y.; et al. Hedgehog-YAP Signaling Pathway Regulates Glutaminolysis to Control Activation of Hepatic Stellate Cells. Gastroenterology 2018, 154, 1465–1479.e13.
- Yu, H.X.; Yao, Y.; Bu, F.T.; Chen, Y.; Wu, Y.T.; Yang, Y.; Chen, X.; Zhu, Y.; Wang, Q.; Pan, X.Y.; et al. Blockade of YAP alleviates hepatic fibrosis through accelerating apoptosis and reversion of activated hepatic stellate cells. Mol. Immunol. 2019, 107, 29–40.
- Alsamman, S.; Christenson, S.A.; Yu, A.; Ayad, N.M.E.; Mooring, M.S.; Segal, J.M.; Hu, J.K.; Schaub, J.R.; Ho, S.S.; Rao, V.; et al. Targeting acid ceramidase inhibits YAP/TAZ signaling to reduce fibrosis in mice. Sci. Transl. Med. 2020, 12, eaay8798.
- Liang, M.; Yu, M.; Xia, R.; Song, K.; Wang, J.; Luo, J.; Chen, G.; Cheng, J. Yap/Taz Deletion in Gli(+) Cell-Derived Myofibroblasts Attenuates Fibrosis. J. Am. Soc. Nephrol. 2017, 28, 3278–3290.
- Gui, Y.; Li, J.; Lu, Q.; Feng, Y.; Wang, M.; He, W.; Yang, J.; Dai, C. Yap/Taz mediates mTORC2-stimulated fibroblast activation and kidney fibrosis. J. Biol. Chem. 2018, 293, 16364–16375.
- Szeto, S.G.; Narimatsu, M.; Lu, M.; He, X.; Sidiqi, A.M.; Tolosa, M.F.; Chan, L.; De Freitas, K.; Bialik, J.F.; Majumder, S.; et al. YAP/TAZ Are Mechanoregulators of TGF-beta-Smad Signaling and Renal Fibrogenesis. J. Am. Soc. Nephrol. 2016, 27, 3117–3128.
- Mia, M.M.; Cibi, D.M.; Binte Abdul Ghani, S.A.; Singh, A.; Tee, N.; Sivakumar, V.; Bogireddi, H.; Cook, S.A.; Mao, J.; Singh, M.K. Loss of Yap/taz in cardiac fibroblasts attenuates adverse remodeling and improves cardiac function. Cardiovasc. Res. 2022, 118, 1785–1804.
- Francisco, J.; Zhang, Y.; Jeong, J.I.; Mizushima, W.; Ikeda, S.; Ivessa, A.; Oka, S.; Zhai, P.; Tallquist, M.D.; Del Re, D.P. Blockade of Fibroblast YAP Attenuates Cardiac Fibrosis and Dysfunction Through MRTF-A Inhibition. JACC Basic Transl. Sci. 2020, 5, 931–945.
- Leach, J.P.; Heallen, T.; Zhang, M.; Rahmani, M.; Morikawa, Y.; Hill, M.C.; Segura, A.; Willerson, J.T.; Martin, J.F. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 2017, 550, 260–264.
- Halder, G.; Johnson, R.L. Hippo signaling: Growth control and beyond. Development 2011, 138, 9–22.
- Zhao, B.; Tumaneng, K.; Guan, K.L. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 2011, 13, 877–883.
- Mia, M.M.; Singh, M.K. The Hippo Signaling Pathway in Cardiac Development and Diseases. Front. Cell Dev. Biol. 2019, 7, 211.
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828.
- Zhao, R.; Fallon, T.R.; Saladi, S.V.; Pardo-Saganta, A.; Villoria, J.; Mou, H.; Vinarsky, V.; Gonzalez-Celeiro, M.; Nunna, N.; Hariri, L.P.; et al. Yap tunes airway epithelial size and architecture by regulating the identity, maintenance, and self-renewal of stem cells. Dev. Cell. 2014, 30, 151–165.
- Szymaniak, A.D.; Mahoney, J.E.; Cardoso, W.V.; Varelas, X. Crumbs3-Mediated Polarity Directs Airway Epithelial Cell Fate through the Hippo Pathway Effector Yap. Dev. Cell. 2015, 34, 283–296.
- Landry, N.M.; Rattan, S.G.; Filomeno, K.L.; Meier, T.W.; Meier, S.C.; Foran, S.J.; Meier, C.F.; Koleini, N.; Fandrich, R.R.; Kardami, E.; et al. SKI activates the Hippo pathway via LIMD1 to inhibit cardiac fibroblast activation. Basic Res. Cardiol. 2021, 116, 25.
- Francisco, J.; Zhang, Y.; Nakada, Y.; Jeong, J.I.; Huang, C.Y.; Ivessa, A.; Oka, S.; Babu, G.J.; Del Re, D.P. AAV-mediated YAP expression in cardiac fibroblasts promotes inflammation and increases fibrosis. Sci. Rep. 2021, 11, 10553.
- Mia, M.M.; Cibi, D.M.; Abdul Ghani, S.A.B.; Song, W.; Tee, N.; Ghosh, S.; Mao, J.; Olson, E.N.; Singh, M.K. YAP/TAZ deficiency reprograms macrophage phenotype and improves infarct healing and cardiac function after myocardial infarction. PLoS Biol. 2020, 18, e3000941.
- Song, K.; Kwon, H.; Han, C.; Chen, W.; Zhang, J.; Ma, W.; Dash, S.; Gandhi, C.R.; Wu, T. Yes-Associated Protein in Kupffer Cells Enhances the Production of Proinflammatory Cytokines and Promotes the Development of Nonalcoholic Steatohepatitis. Hepatology 2020, 72, 72–87.
- Anorga, S.; Overstreet, J.M.; Falke, L.L.; Tang, J.; Goldschmeding, R.G.; Higgins, P.J.; Samarakoon, R. Deregulation of Hippo-TAZ pathway during renal injury confers a fibrotic maladaptive phenotype. FASEB J. 2018, 32, 2644–2657.
- Chen, J.; You, H.; Li, Y.; Xu, Y.; He, Q.; Harris, R.C. EGF Receptor-Dependent YAP Activation Is Important for Renal Recovery from AKI. J. Am. Soc. Nephrol. 2018, 29, 2372–2385.
- Chen, J.; Wang, X.; He, Q.; Bulus, N.; Fogo, A.B.; Zhang, M.Z.; Harris, R.C. YAP Activation in Renal Proximal Tubule Cells Drives Diabetic Renal Interstitial Fibrogenesis. Diabetes 2020, 69, 2446–2457.
- Mia, M.M.; Singh, M.K. Emerging roles of the Hippo signaling pathway in modulating immune response and inflammation-driven tissue repair and remodeling. FEBS J. 2022; in press.
- Xiao, Y.; Hill, M.C.; Li, L.; Deshmukh, V.; Martin, T.J.; Wang, J.; Martin, J.F. Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes Dev. 2019, 33, 1491–1505.
- Leung, J.Y.; Wilson, H.L.; Voltzke, K.J.; Williams, L.A.; Lee, H.J.; Wobker, S.E.; Kim, W.Y. Sav1 Loss Induces Senescence and Stat3 Activation Coinciding with Tubulointerstitial Fibrosis. Mol. Cell. Biol. 2017, 37, e00565-16.
- Shao, D.; Zhai, P.; Hu, C.; Mukai, R.; Sciarretta, S.; Del Re, D.; Sadoshima, J. Lats2 promotes heart failure by stimulating p53-mediated apoptosis during pressure overload. Sci. Rep. 2021, 11, 23469.
- Gokey, J.J.; Sridharan, A.; Xu, Y.; Green, J.; Carraro, G.; Stripp, B.R.; Perl, A.T.; Whitsett, J.A. Active epithelial Hippo signaling in idiopathic pulmonary fibrosis. JCI Insight 2018, 3, e98738.
- Triastuti, E.; Nugroho, A.B.; Zi, M.; Prehar, S.; Kohar, Y.S.; Bui, T.A.; Stafford, N.; Cartwright, E.J.; Abraham, S.; Oceandy, D. Pharmacological inhibition of Hippo pathway, with the novel kinase inhibitor XMU-MP-1, protects the heart against adverse effects during pressure overload. Br. J. Pharmacol. 2019, 176, 3956–3971.
- He, X.; Tolosa, M.F.; Zhang, T.; Goru, S.K.; Ulloa Severino, L.; Misra, P.S.; McEvoy, C.M.; Caldwell, L.; Szeto, S.G.; Gao, F.; et al. Myofibroblast YAP/TAZ activation is a key step in organ fibrogenesis. JCI Insight 2022, 7, e146243.
- Caliari, S.R.; Perepelyuk, M.; Cosgrove, B.D.; Tsai, S.J.; Lee, G.Y.; Mauck, R.L.; Wells, R.G.; Burdick, J.A. Stiffening hydrogels for investigating the dynamics of hepatic stellate cell mechanotransduction during myofibroblast activation. Sci. Rep. 2016, 6, 21387.
- Sharifi-Sanjani, M.; Berman, M.; Goncharov, D.; Alhamaydeh, M.; Avolio, T.G.; Baust, J.; Chang, B.; Kobir, A.; Ross, M.; St Croix, C.; et al. Yes-Associated Protein (Yap) Is Up-Regulated in Heart Failure and Promotes Cardiac Fibroblast Proliferation. Int. J. Mol. Sci. 2021, 22, 6164.
- Zhang, J.; Liu, J.; Gao, S.; Lin, W.; Gao, P.; Gao, K.; Zhang, Y.; Du, K.; Yang, X.; Wang, W.; et al. Antiapoptosis and Antifibrosis Effects of Qishen Granules on Heart Failure Rats via Hippo Pathway. Biomed. Res. Int. 2019, 2019, 1642575.
- Wu, P.; Liu, Z.; Zhao, T.; Xia, F.; Gong, L.; Zheng, Z.; Chen, Z.; Yang, T.; Duan, Q. Lovastatin attenuates angiotensin II induced cardiovascular fibrosis through the suppression of YAP/TAZ signaling. Biochem. Biophys. Res. Commun. 2019, 512, 736–741.
- Liu, M.; Liu, S.; Tan, W.; Tang, F.; Long, J.; Li, Z.; Liang, B.; Chu, C.; Yang, J. Gaseous signalling molecule SO2 via HippoMST pathway to improve myocardial fibrosis of diabetic rats. Mol. Med. Rep. 2017, 16, 8953–8963.
- Ramjee, V.; Li, D.; Manderfield, L.J.; Liu, F.; Engleka, K.A.; Aghajanian, H.; Rodell, C.B.; Lu, W.; Ho, V.; Wang, T.; et al. Epicardial YAP/TAZ orchestrate an immunosuppressive response following myocardial infarction. J. Clin. Investig. 2017, 127, 899–911.
- Del Re, D.P.; Matsuda, T.; Zhai, P.; Gao, S.; Clark, G.J.; Van Der Weyden, L.; Sadoshima, J. Proapoptotic Rassf1A/Mst1 signaling in cardiac fibroblasts is protective against pressure overload in mice. J. Clin. Investig. 2010, 120, 3555–3567.
- Mia, M.M.; Chelakkot-Govindalayathil, A.L.; Singh, M.K. Targeting NF2-Hippo/Yap signaling pathway for cardioprotection after ischemia/reperfusion injury. Ann. Transl. Med. 2016, 4, 545.
More