Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Bingran Yu and Version 2 by Amina Yu.
Antibiotic resistance has posed a great threat to human health. The emergence of antibiotic resistance has always outpaced the development of new antibiotics, and the investment in the development of new antibiotics is diminishing. Supramolecular self-assembly of the conventional antibacterial agents has been proved to be a promising and versatile strategy to tackle the serious problem of antibiotic resistance.
- supramolecular assembly
- antibacteria
1. Introduction
Antibiotic resistance is a growing problem that causes 700,000 deaths per year worldwide, and a recent prediction indicates that bacterial infections will cause 10 million deaths by 2050 [1][2][3][4][5][6][1,2,3,4,5,6]. The rapidly growing antibiotic resistance is mainly driven by misuse of antibiotics in human medicine and abuse of antibiotics in animal medicine and husbandry [7][8][9][10][7,8,9,10]. The emergence of multi-drug-resistant infections makes the situation worse, as multi-drug-resistant infections require prolonged and high-cost antibiotic therapy [11][12][13][14][11,12,13,14]. However, the investment in research and development of new antibiotics is diminishing, as the emergence of resistance has outpaced the development of new antibiotics [15][16][17][18][15,16,17,18]. Therefore, developing novel approaches to tackle the serious antibiotic resistance crisis is urgently needed.
Researchers in relevant fields are making great efforts to discover new drugs and develop new antibacterial strategies and technologies to address the serious issues caused by antibiotic resistance [19][20][21][22][23][24][25][26][19,20,21,22,23,24,25,26]. Supramolecular self-assembly focuses on the autonomous organization of components into multi-component systems through intermolecular noncovalent interactions, including electrostatic, hydrophobic, hydrogen-bonding, metal-ligand coordination, charge–transfer, van der Waals, and π-π stacking interactions [27][28][29][30][31][32][27,28,29,30,31,32]. The dynamic properties and integration features of supramolecular self-assembly endow the assembly with extraordinary functions, which cannot be empowered by traditional covalent modification strategies. Therefore, supramolecular self-assembly strategies have demonstrated great potential applications in the biomaterial field. In particular, supramolecular self-assembly strategies have been applied in combating bacteria and bacterial biofilms.
2. Supramolecular Self-Assembly of Antibiotics
Since their introduction in the 1940s, antibiotics have been heavily applied to treat infectious diseases. After the “Golden Age” of antibiotics, semisynthetic and chemically modified antibiotics have been deemed as the bright way to develop new antibiotics [33][34][35][36][37][33,34,35,36,37]. However, these innovative compounds cannot address the antibiotic-resistant issues as bacteria could rapidly acquire tolerance after a period of gene response. Thus, developing a novel antibacterial strategy based on the existing antibiotics to combat resistance is greatly needed. Supramolecular self-assembly of antibiotics is a promising strategy to improve therapeutic efficiency, reduce nonspecific cytotoxicity, and depress drug resistance to antibiotics. Bhosale and coworkers have summarized the supramolecular self-assembly of amino-glycoside antibiotics [38][38], and for readers interested in this aspect, please refer to the review.
The host–guest complexations between antibiotics and macrocyclic hosts are highly potential approaches to enhance antibiotics activity. Sinisterra et al. reported that the complexation of doxycycline (Dox) and β-cyclodextrin (β-CD) showed greater antimicrobial activity than free doxycycline (Figure 1) [39]. Isothermal titration calorimetry (ITC) data indicated that the free doxycycline and the Dox/β-CD complexation bind with the cell membrane through different interactions. The interaction of free doxycycline with the cell membrane is an ion-paring and hydrogen bond, while the interaction of Dox/β-CD complexation with the cell membrane is a much stronger hydrogen bond. The Dox/β-CD complexation could serve as a local and sustained source to improve treatment efficacy. Dend et al. also reported the complexation of per-6(4-methoxylbenzyl)-amino-6-deoxy-β-cyclodextrin (pMBA-βCD) and methicillin (Met) to increase the water solubility of Met and its antibacterial activities against methicillin-resistant Staphylococcus aureus (MRSA) [40], which was presumed that the Met/pMBA-βCD complex improved the affinity between the active β-lactam moiety and the narrow active site groove of MRSA PBP2a, and facilitate the acylation [41][42][41,42] (Figure 1). The solubility and antibacterial activity of ciprofloxacin were also enhanced by Mono-6-deoxy-6-aminoethylamino-β-cyclodextrin (mET-βCD, Figure 1) [43]. NOESY NMR indicated that the ethylenediamine moiety of mET-βCD induces stable hydrogen bonding with primary hydroxyls of β CD, leading to the formation of this oval-shaped cavity, which could encapsulate quinolone and the cyclopropyl groups of ciprofloxacin. D-mannose and D-glucose-grafted cyclodextrins were also developed as “Trojan Horse”-like nanocarriers for loading hydrophobic antibiotics (ciprofloxacin, erythromycin, and rifampicin) and potentiate their activity, as the functionalized sugars on CD are chemoattractant for the bacteria and could promote uptake (Figure 1) [44].
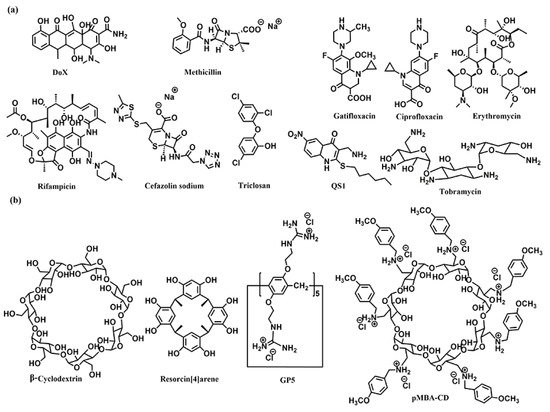
Figure 1. Chemical structures of (a) the antibiotics, and (b) the co-assembly materials used for fabricating supramolecular self-assembly of antibiotics.
Kumari et al. also fabricated the host–guest complexation between resorcin[4]arene and gatifloxacin to improve antibacterial activity [45]. These host–guest complexations indicated that strategies based on host–guest interactions are highly potential approaches to constructing novel antimicrobial formulations, which would improve antibacterial activity, water-solubility, and biocompatibility of traditional antibiotics.
Microbial biofilms showed restricted drug accessibility and antibiotic resistance, and most cells in the biofilms changed their metabolic mode to a dormant state. All of these factors result in great challenges in disrupting biofilms [46][47][46,47]. Supramolecular self-assembly of antibiotics is also a potential strategy to disrupt biofilms. Wang and coworkers also developed host–guest complexation between guanidinium per-functionalized pillar[5]arene (GP5) and the antibiotic cefazolin sodium to synergistically eradicate Escherichia coli (E. coli) biofilm [48]. In vitro experiment showed that GP5 could disrupt biofilm, as guanidinium per-functionalized pillar[5]arene could penetrate through biofilm barriers and destroy biofilm-enclosed bacteria. After GP5 formed the host–guest complex with cefazolin sodium, the host–guest complex penetrated through biofilm due to the high penetrability of preorganized multiple guanidiniums on pillar[5]arene skeleton (Figure 2). The GP5 and cefazolin sodium, therefore, could effectively breach the bacterial membranes and work synergistically to kill the bacteria within the biofilm matrix.
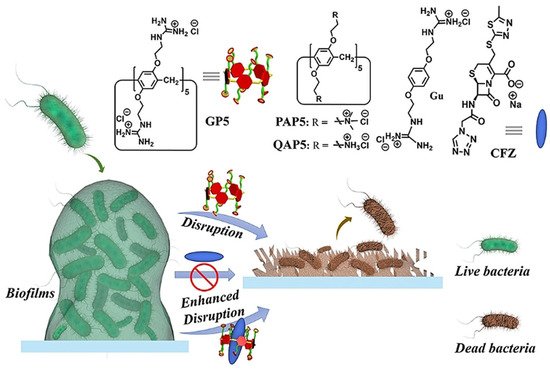
Figure 2. Disruption of biofilms by GP5 and GP5⊃CFZ. Reprinted with permission from Ref. [48]. Copyright 2021 Wiley-VCH.
Encapsulation and delivery of antibiotics through the self-assembly of lipids is a promising way to enhance the bioavailability of antibiotics [49]. Macrophage membrane encapsulated antibiotic was construed to selectively enter into the infected macrophages and efficiently kill intracellular bacteria [50]. The hydrophobic triclosan and hydrophilic ciprofloxacin were covalently conjugated together, and the obtained amphiphilic prodrug self-assembled into nanoparticles. With further encapsulation with macrophage membranes, the nanoparticles showed similar Toll-like receptor expression and negative surface charge as their precursor murine macrophage/human monocyte cell lines. Such features allowed uptake of the infected macrophages/monocytes through positively charged, lysozyme-rich membrane scars created during staphylococcal engulfment.
Combined antibiotic administration based on co-assembly is also an effective way to eliminate resistant infections. Lehr et al. developed a co-assembled nanoparticle of synthesized amphiphilic lipid (squalenyl hydrogen sulfate), hydrophilic antibiotic tobramycin, and a quorum sensing inhibitor (QS1) to synergistically eradicate Pseudomonas aeruginosa (P. aeruginosa) biofilm [51].
Wang and coworkers fabricated cascade-targeting poly(amino acid) nanoparticles to sequentially target macrophages and intracellular bacteria and fulfill on-site antibiotic delivery. The mannose-decorated poly(α-N-acryloylphenylalanine)-block-poly(β-N-acryloyl-d-aminoalanine) could self-assemble into nanoparticles and efficiently load rifampicin due to abundant noncovalent interactions between the drug and polymer backbone (Figure 3). The mannose groups from the surface of the nanoparticles promote macrophage-specific uptake and intracellular accumulation. After exposing the D-aminoalanine moieties in the acidic phagolysosome, the nanoparticles escape from lysosomes and target intracellular bacteria through peptidoglycan-specific binding. Subsequently, rifampicin was precisely released regardless of the states and locations of the intracellular MRSA [52].
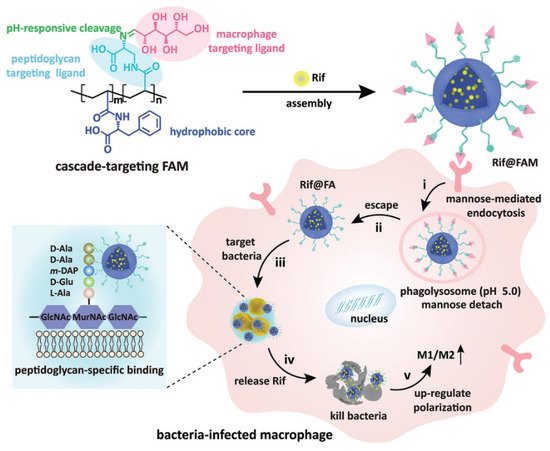
Figure 3. The cascade-targeting DDS eliminates intracellular MRSA. Reprinted with permission from Ref. [52]. Copyright 2022 Wiley-VCH.
3. Supramolecular Self-Assembly of AMPs
Antimicrobial peptides (AMPs), a group of functional peptides that display antimicrobial activities, are potential therapeutic agents due to their broad-spectrum antibacterial activities and low degrees of antimicrobial resistance [53][54][55][53,54,55]. As peptides are outstanding and feasible building blocks for self-assembly, the artificial peptides with self-assembling capacities have been applied in antibiotic delivery, antibacterial surface defense, bio-sensing of bacteria, and as bacteriostatic agents [56][57][58][56,57,58]. The self-assembly of AMPs could not only enhance antimicrobial efficacy by locally enhancing the antimicrobial peptide concentration on the bacterial surface but also promote the biological stability of the fragile peptide.
AMPs are drawing great attention in modern medicine due to their high biological activity. Their low stability leads to undesirable bioavailability, as these agents could be degraded by proteases. Further, systemic toxicity is also a major issue for some classes of AMPs. Recently, Li et al. exploited the host–guest complexion between and pexiganan (PXG) to decrease hemolysis and improve the stability of PXG [59]. The two large-sized macrocycles showed strong interactions towards PXG, giving association constants in the magnitude of 104 and 105 M−1. The host–guest complexation remarkably improves metabolic stability under endogenous proteases conditions and decreases hemolysis of PXG.
Lin et al. developed a supramolecular trap to boost the low-dose antibacterial activity of colistin against E. coli by preventing the interaction between colistin and free LPS [60]. The molecular trap was fabricated from a subnanometer gold nanosheet with methyl motifs (SAuM) and could directly target and capture free LPS by binding to and compressing the packing density of lipid A, thus reducing the risk of endotoxin-induced sepsis and preventing its interaction with colistin (Figure 4). The binding specificity assay results showed that SAuM is a strong competitor of colistin for binding with free LPS. The higher driving force of SAuM to bind with LPS is mainly due to the fact that the sheet-like structure of SAuM can precisely bind the lipid A of LPS to form a steric wall that efficiently inhibits colistin binding. This supramolecular trap allows the therapeutic window of colistin to be expanded to low-dose concentrations for the treatment of Gram-negative bacteria infections while also minimizing the risk of endotoxemia.
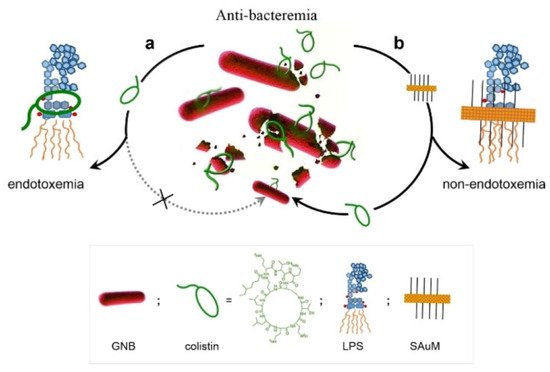
Figure 4. A Supramolecular Trap to Increase the Antibacterial Activity of Colistin. In Path a, free LPS can block the function of colistin. And the free LPS can be sequestered by SAuM in the circulating blood (in Path b), thus promoting the killing efficiency of colistin against GNB while also minimizing endotoxemia. Reprinted with permission from Ref. [60]. Copyright 2020, Wiley-VCH.
Tang and coworkers developed a novel peptide-based co-assembled hydrogel to effectively and specifically capture and kill MRSA bacteria. The supramolecular self-assembly of 9-fluorenylmethyloxycarbonyl-L-phenylalanin (Fmoc-L-Phe) and amino-acid-modified conjugated oligomer OTE-D-Phe formed a new and biocompatible low-molecular weight hydrogel [61]. The hydrogel was composed of a thick and rough fibrous network, which could spontaneously capture MRSA and E. coli efficiently. The hydrogel exhibited efficient and specific antibacterial activity against MRSA due to specific interaction with lipid domains of the MRSA membrane.