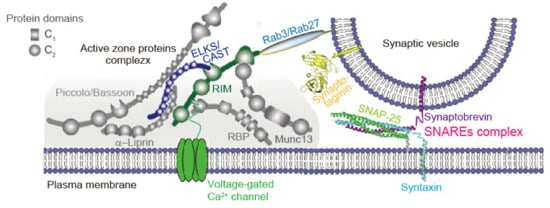
2. Structure and Function of Presynaptic Neurotransmitter Release Sites
Within a synapse, neurotransmitter release is restricted to specialized presynaptic structures called AZs
[4][8][4,8]. The AZ is a highly organized structure composed of sophisticated protein machinery (
Figure 1)
[1][2][3][1,2,3]. The AZ serves as a platform for SV exocytosis. Synaptotagmins, Ca
2+ sensor proteins expressed on the SV membrane, mediate SV exocytosis cooperating with the SNAREs complexes, fusion machinery proteins, near Ca
V channels (
Figure 1). The tight spatial organization enables to induce the synchronous SV fusion upon Ca
2+ entry, and sets the synaptic strength
[9][20].
2.1. AZ Structure
AZ proteins, including Munc13, RIM, RIM-BP, CAST/ELKS, Bassoon, Piccolo, and Liprin-α
[10][11][12][13][14][15][16][17][21,22,23,24,25,26,27,28], are all relatively large proteins, and form a large macromolecular complex interacting with each other via significant domain structures (
Figure 1)
[1]. RIM (Rab3-interacting molecules)
[17][28] and RIM-BP (RIM-binding protein)
[18][29] are essential for the inclusion of Ca
V channels
[19][20][30,31]. A molecular complex consisting of RIM and the C-terminal tails of the Ca
V channels, which determines the recruitment of Ca
V2 channels to the AZ, includes RIM-BP and CAST (cytomatrix at the active zone-associated structural protein)
[13][24]/ELKS
[21][32]. In contrast, interaction of RIM, RIM-BP, and CAST/ELKS may be essential for the constitution of the AZ structure: RIM and ELKS double deletion induces the loss of Munc13-1, Bassoon, Piccolo, RIM-BP2, and the Ca
V2 channels
[3]. CAST controls Ca
V channel density
[22][33], AZ size
[23][24][34,35], and SV docking
[25][15]. The disruption of CAST interaction with RIM impairs synaptic transmission
[14][25]. Among four Liprin-α, Liprin-3α, which is strongly expressed in the brain, has recently been reported to control the co-recruitment of RIM and Mun 13 via protein kinase C (PKC)-mediated phosphorylation
[26][36].
Figure 1. Presynaptic active zone assembly model and synaptic vesicle fusion machinery proteins. Diagram represents active zone (AZ) proteins’ complex in a liquid droplet (gray) and a docked synaptic vesicle (SV). A recent in vitro study indicates that SVs coat the surface of condensed liquid droplets
[27][37]. In the droplet, the AZ is a highly organized structure that recruits voltage-gated Ca
2+ channel and docks SV. Reproduced with permission from ref.
[3]. Copyright 2016, Wang et al. Priming factors promote proper assembly consisting of the pre-fusion state of the fusion machinery protein complex and SNAREs/synaptotagmin/complexin (not shown)
[28][38]. The tight spatial organization ensures fast exocytosis upon Ca
2+ entry, and it provides molecular machinery to set and regulate synaptic strength, presynaptic short-term plasticity, and homeostatic synaptic plasticity. For detailed order of events of vesicle tethering, docking and fusion, please refer to reviews
[29][30][11,39].
2.2. AZ Proteins Control SV States
The AZ proteins play a central role in neurotransmitter release by localizing Ca
V channels in the release sites as discussed above, and also in setting the defined states of SVs termed as tethering, docking, priming, and fusion
[1]. Bassoon and Piccolo mediate SV tethering and regulate SV reloading
[31][32][33][40,41,42]. RIM seems to be a key protein for SV dynamics
[34][35][43,44]. RIM mediates the linkage of docked SV via Rab3/Rab27, synaptic vesicle proteins and members of the family of low-molecular weight guanosine triphosphatases (GTPases)
[30][36][37][39,45,46], and Ca
V channels in the neurotransmitter release site
[38][47] (
Figure 1). Removing RIM removes SV docking and slows down the exocytosis speed
[3]. RIM also mediates SV priming
[39][40][48,49], leading to a SV that can rapidly fuse upon Ca
2+ stimulation
[30][39]. SV priming is also mediated by ELKS, RIM-BP, and Munc13
[39][40][41][42][48,49,50,51]. In addition, the involvement of Mover, another AZ protein, in the super-priming of SVs, was recently reported
[43][52]. Munc13 and Munc18 interact with the fusion machinery and regulate exocytosis
[44][45][46][47][48][53,54,55,56,57] (see
Section 3.3). For more complete reviews on molecular mechanisms, please refer to recent articles
[4][8][4,8].
2.3. AZ Protein Complex Formation
To explore AZ assembly models AZ protein interactions have long been examined (
Figure 1)
[1][3][1,3]. Except for RIM and RIM-BP, which are required for Ca
V channel clustering
[19][49][30,58], however, deletion of single-AZ proteins causes only mild effects on the AZ structure, suggesting that individual protein interactions unlikely have a major role in the AZ assembly
[8]. A recent in vitro study has proposed a new model whereby AZ assembly relies on liquid–liquid phase separation principles
[50][59]. RIM, RIM-BP, and Ca
V channels form dense clusters on the supported lipid membrane bilayers via phase separation. Liprin-3α, which co-recruits RIM and Mun 13 via PKC-mediated phosphorylation, also undergoes phase separation in transfected HEK cells
[26][36]. The new model suggests that multiple low-affinity interactions very likely drive AZ formation
[8], as the suggested models of droplet-like condensate formation with protein interactions in clustering of SVs
[51][52][60,61] and postsynaptic density
[53][62]. Liquid–liquid phase separation or low-affinity interaction can contribute to membrane anchoring
[27][51][37,60].
2.4. A Possible Model for SV Pools Formation
In presynaptic terminals or boutons four functional pools of SVs are organized: the readily releasable pool (RRP), the recycling pool, and the reserve and resting pools
[5][54][5,63]. To maintain sustainable neurotransmitter release, the RRP has to be replenished constantly with SVs. During prolonged neuronal activity the replenishment is achieved by either the rapid reuse of fused vesicles, or the recruitment of new SVs from the reserve pool
[5]. All SVs which participate in activity-induced neurotransmitter release comprise the recycling pool
[54][63].
The AZ proteins RIM, RIM-BP, and ELKS form condensed liquid droplets via phase separation. SVs from rat brains and small unilamellar vesicles (SUVs) coat the surfaces of the condensed liquid droplets
[27][37]. The SUV-coated RIM/RIM-BP condensates enable to cluster Ca
V channels anchored on membranes. Strikingly, synapsin/SUV condensates wrap SUV-coated RIM/RIM-BP condensates. The formation of two distinct SUV pools is likely the reserve and tethered SV pools in presynaptic boutons
[27][37]. These studies provide a possible model for SV pools formation reconstituting a presynaptic bouton-like structure that mimics the RRP with the SV-tethered AZ and the connected reserved pool with the synapsin-clustered SV condensates.
2.5. AZ Assembly Stability
Bassoon and Piccolo maintain synapse integrity by regulating protein ubiquitination and degradation. The aberrant degradation of multiple presynaptic proteins is induced by loss of Bassoon and Piccolo. Loss of Bassoon and Piccolo also lead to synapse degeneration, mediated in part by the E3 ubiquitin ligase Siah1 that is an interacting partner of Bassoon and Piccolo
[55][64]. In boutons lacking Bassoon and Piccolo the destruction of SVs is associated with presynaptic autophagy, a process dependent on poly-ubiquitination. The gain or loss of function of Bassoon alone suppressed or enhanced presynaptic autophagy, respectively, indicating a fundamental role for Bassoon in the local regulation of presynaptic autophagy
[32][41]. Bassoon and Piccolo are critical regulators for stabilizing the AZ assembly by inhibiting degradation.
3. Synaptic Vesicle Exocytosis
SV fusion is a series of events, and its flow is controlled by a number of protein–protein interactions: Fusogenic SNAREs, the Ca
2+-sensor synaptotagmin, the activator/regulator complexin, the assembly factors Munc18 and Munc13, and the disassembly factors NSF and SNAP have been identified as key factors of the core synaptic fusion machinery
[29][11].
3.1. SV Fusion Complex—A Model where Ca
2+
Releases Inhibition of SV Fusion
Synaptotagmins expressed on the SV membrane cooperating with the SNAREs complex has been implicated to mediate the fusion of neurotransmitter-containing SVs with the presynaptic plasma membrane
[1][2][1,2]. Recent structural and functional studies suggest that AP-evoked sub-millisecond SV fusion occurs with the release of inhibition by Ca
2+ binding to synaptotagmin
[28][56][38,65]. Complexin, a cytoplasmic protein that is crucial for the regulation of neurotransmitter release
[57][58][66,67], contributes to the inhibition process. The structure of SNAREs, synaptotagmin-1, and complexin-1 complex clarifies two interfaces: the primary interface between synaptotagmin-1 and SNAREs, and a tripartite interface between SNAREs, complexin-1, and synaptotagmin-1. These two interfaces of the complex suggest the cooperation of all three components in the evoked SV fusion
[29][11]. The tripartite complex of SNAREs/complexin/synaptitagmin-1 is formed in the absence of Ca
2+, suggesting a prefusion complex inhibiting SV fusion
[29][11].
Synaptotagmins contain cytoplasmic C2A and C2B domains
[59][60][68,69]. The structure of SNAREs, complexin-1, and synaptotagmin-1 complex clarifies a tripartite interface between one synaptotagmin-1 C2B domain, SNAREs and complexin-1
[56][65]. This tripartite interface formation is promoted by complexin-1 binding to SNAREs. The second synaptotagmin-1 C2B domain simultaneously interact with the other side of the SNAREs via a pairwise interface. Synaptotagmin-1 C2B residues is responsible for the Ca
2+-triggered synchronous neurotransmitter release and the suppression of spontaneous release. Synaptotagmin-1 C2B residues are involved in both the tripartite interface of SNAREs, complexin-1 and synaptotagmin-1 and the primary interfaces of SNAREs and synaptotagmin-1
[29][56][61][11,65,70].
Synaptotagmin-2, at the calyx of the Held synapse and in some GABAergic neurons, regulates neurotransmission redundantly with synaptotagmin-1
[62][63][71,72]. Synaptic transmission depends on synaptotagmin-1 in the early postnatal calyx of the Held synapses, but later switches to synaptotagmin-2
[63][72], suggesting dynamic changes in synaptotagmin content at the synapses during development. In the hippocampal neurons, synaptotagmin-7 acts redundantly with synaptotagmin-1 in the maintenance of the RRP of SVs
[64][73]. The loss of synaptotagmin-1 function in immature neurons can compensate with the expression of synaptotagmin-7
[65][12]. The structural study of the tripartite complex of SNAREs/complexin/synaptitagmin-1 has not yet been applied for synaptotagmin-2 or -7 in the fusion machinery.
3.2. Asynchronous SV Fusion
Ca
2+-triggered evoked SV fusion causes synchronous neurotransmitter release, which occurs within tens of microseconds of a stimulus and completes within several milliseconds
[66][74], transmitting a fast and reliable signal. The evoked SV fusion also causes asynchronous neurotransmitter release, which sets in more slowly and can persist for tens or hundreds of milliseconds
[67][68][75,76], and has influences on network parameters, including the efficacies of neurotransmission, synchronicity, and plasticity
[69][70][71][77,78,79]. Synaptotagmin-7
[64][71][73,79] and Doc2
[72][73][80,81] are reported as being high-affinity Ca
2+ sensors for asynchronous release. Both Doc2 and synaptotagmin-7 exhibit the slow Ca
2+-regulated membrane-binding kinetics and high affinities required for asynchronous release
[72][74][75][80,82,83].
Synaptotagmin-7 mediates asynchronous release in cultured hippocampal neurons
[64][73], and in granule cell synapses as well as in inhibitory synapses formed between basket cells and Purkinje neurons in the cerebellum
[76][77][84,85], and in excitatory neocortex synapses formed between pyramidal cells and Martinotti neurons
[78][86]. The role of synaptotagmin-7 in asynchronous release is usually only apparent when more than one stimulus is applied
[64][77][73,85]. At the neuromuscular junction of
Caenorhabditis elegans, synaptotagmin-3 triggers delayed Ca
2+-dependent neurotransmitter release following fast synaptotagmin-1-mediated release. The fast and slow properties of neurotransmitter release are due to essentially different C2 domains in synaptotagmin-1 and -3
[79][87].
Synaptotagmin-7 underlies phasic somatodendritic dopamine release and its Ca
2+ sensitivity in the substantia nigra pars compacta. In contrast, synaptotagmin-1, underlying axonal dopamine release, plays a role in tonic dopamine release. However, synaptotagmi-1 can facilitate phasic dopamine release after synaptotagmin-7 deletion
[80][88]. These results indicate that synaptotagmin Ca
2+ sensors subserve different aspects of the transmitter release processes.
Doc2α mediates asynchronous release in cultured hippocampal neurons, both after single AP and during AP trains
[72][75][81][82][80,83,89,90]. Loss of asynchronous release in Doc2α deficient neurons can be rescued by Doc2β
[72][80]. Inconsistently, Purkinje cells to deep cerebellar nuclei synapses
[83][91], nor autaptic cultured hippocampal neurons lacking Doc2 shows no significant changes in asynchronous release
[73][81]. At excitatory synapses in mouse hippocampus, the major Ca
2+ sensor for asynchronous release is Doc2α, while synaptotagmin-7 supports this process through the activity-dependent docking of SVs
[84][92].
3.3. Regulation of the Prefusion Complex
Munc18 binds to free syntaxin-1A, and the heterodimeric complex prevents the ternary SNAREs formation
[85][86][93,94]. The syntaxin/Munc18 complex is forwarded to the ternary trans-SNAREs catalyzed by Munc13
[45][87][88][54,95,96]. Munc13 promotes the assembly of the SNAREs in cooperation with Munc18, forming the parallel configuration of all components of the SNAREs
[41][50], suggesting that Munc13 and Munc18 are assembly factors for establishing the ternary SNAREs. Both syntaxin-1A (membrane SNARE) and synaptobrevin-2 (vesicle SNARE) weakly interact with the MUN domain of Munc13
[41][46][89][90][50,55,97,98]. An interaction between synaptobrevin-2 in the membrane proximal region and Munc13 in the MUN domain is essential for the function of Munc13
[46][55]. The efficiency of Ca
2+-triggered SV fusion is significantly increased by Munc18 and Munc13 in a reconstituted fusion assay
[41][50].
3.4. Disassembly of the Postfusion SNAREs
After SV fusion, the ternary SNARE complex is disassembled for recycling the individual SNARE proteins. The ATPase NSF disassembles the ternary SNARE complex, with ATP hydrolysis cooperating with the adaptor protein, SNAP
[91][92][93][99,100,101]. These catalyzing molecules of NSF and SNAPs interacting with ternary SNAREs form the so-called 20S complex, for starting state of the disassembly process. However, the molecular mechanism for NSF-mediated SNARE complex disassembly is not yet clarified
[29][11].
High-resolution reconstruction of the NSF, αSNAP, and the full-length soluble neuronal SNARE complex (composed of syntaxin-1A, synaptobrevin-2, SNAP-25A) demonstrated the molecular interactions between NSF and αSNAPs with the SNAREs as follows.
[94][102] Electrostatic interactions by which two αSNAP molecules interface with a specific surface of the SNARE complex. This interaction positions the SNAREs such that the 15 N-terminal residues of SNAP-25A are loaded into the ring pore of NSF via a spiral pattern of interactions between a conserved tyrosine NSF residue and SNAP-25A backbone atoms. This loading process likely precedes ATP hydrolysis. Subsequent ATP hydrolysis then drives complete disassembly. Details of the molecular mechanisms for SNARE complex disassembly machinery have been reported in a review
[29][11].
4. Replenishment of Release Site with Synaptic Vesicles
4.1. SV Dynamics after AP
The ‘zap-and-freeze’ method, generating an AP and following high-pressure freezing at defined time points, can enable characterization of the spatial and temporal organization of the SV fusion sites following an AP firing
[95][13]. With this technical approach, SV dynamics were morphologically analyzed at milliseconds time points after AP in presynaptic terminals of mouse hippocampal neurons in culture. 2% of the synaptic profiles in unstimulated synapses showed exocytic pits in the AZ. Within 5 msec after AP, the synchronous fusion of multiple SVs occurred throughout a single AZ. During synchronous fusion, docked SVs reduced by ~40%, in contrast, SVs close to the membrane (between 6 and 10 nm) slightly increased. Such SVs are possibly in a “loose state”, with SNAREs, synaptotagmin-1, and Munc13 still being engaged
[96][103]. At 5 msec, 18% of the synaptic profiles showed exocytic pits in the AZ. By 11 msec, fused SVs collapsed into the plasma membrane. From 5 to 11 msec, asynchronous fusion followed in the center of the AZ. During asynchronous fusion, the docked SVs are not further depleted in spite of their ongoing fusion, suggesting an active recruitment of SVs during this process. At 14 msec, the docked SVs are fully restored to prestimulus levels with newly docked SVs. This fast recovery of docked SVs is Ca
2+-dependent, and temporary lasting for 100 msec or less. During the recovery period, newly docked SVs undock or fuse, indicating that the sequence of rapid redocking and subsequent slow undocking may underlie the synaptic facilitation.
The series of snapshot images taken by the ‘zap-and-freeze’ method demonstrated millisecond SV dynamics, such as synchronous and asynchronous fusion, undocking, and docking, which are regulated by AZ proteins as discussed in
Section 2.2, following transient Ca
2+ elevation, with the opening of Ca
V channels accompanying AP.
4.2. AZ Proteins
AZ proteins contribute to the establishment of multiple functionally definable stages of the SV state, as discussed in
Section 2Section 2.2.
2. For the replenishment of release site with SVs, possible functions of RIM-BP, CAST, Bassoon, and Piccolo have been reported.
RIM-BP, interacting with RIM and Ca
V channels
[49][58], organizes the SV release site topography
[27][51][37,60]. RIM-BP in
Drosophila supports a rate-limiting stage required for the replenishment of high release-probability SVs that follows depletion of SVs
[97][104]. In a mammalian auditory synapse of the cochlear nucleus RIM-BP controls both the release probability and the SV replenishment
[20][31]. Loss of RIM-BP2 lowered the release probability, due to a slowed down of the Ca
2+-dependent fast SV replenishment. The ultrastructural studies revealed reduced docked SVs and proximal SVs, in addition to an impaired Ca
V channels topography in the AZ
[20][31]. Ca
V2.1 channel localization in the AZ is specifically controlled by RIM-BP binding to Bassoon
[98][105]. Thus, it is likely that RIM-BP, interacting with Bassoon via RIM, controls the rate of Ca
2+-dependent fast SV replenishment at the fast central auditory synapse
[20][31].
CAST/ELKS also controls SV replenishment
[25][15]. In cultured sympathetic presynaptic terminals, CAST
S45 is phosphorylated in an activity-dependent manner. Expression of the phosphomimetic-CAST reduced the SV number in the RRP. The paired-AP protocol experiments indicate that the phosphorylation of CAST
S45 causes paired-EPSP depression with a time window of 30–120 msec after the first AP. Overexpression of the phosphonegative-CAST reduced the paired-EPSP depression (<200 msec), suggesting that phosphorylated CAST
S45 downregulates SV reloading shortly after AP, but not over a longer period. The possible kinase is a serine/threonine kinase SAD-B, a presynaptic kinase that is associated with the AZ cytomatrix and SVs, and that phosphorylates CAST
S45 in vitro
[25][15]. Acute deletion of CAST significantly delayed the rate of fast SV reloading, following the RRP depletion with AP bursts. These results indicate that CAST is required for fast SV reloading; however, the phosphorylated CAST, within 200 msec after AP, brakes transmitter release by slowing down SV reloading. The braking of SV reloading may save SVs for an incoming AP to the presynaptic terminal. RIM1, a binding partner of CAST, interacting with Munc13-1, is implicated in SV docking and priming
[99][106]. These protein interactions are likely involved in the CAST-mediated fast replenishment of release sites with release-ready SV.
Bassoon participates in the reloading of SVs to release sites in excitatory synapses of cerebellar mossy fiber connecting to granule cell
[31][40]: Bassoon knockout enhanced short-term synaptic depression during sustained high frequency stimulation, halving the SV reloading rate, whereas it caused no effect on basal synaptic transmission. At the central endbulb synapse in Bassoon knockout mice, SV replenishment rate was slowed down, whereas vesicle number and the accompaniment were normal
[100][19]. These results suggest a role for Bassoon in speeding up high activity-dependent SV tethering, leading to the rapid replenishment of release sites. At the rat calyx of the Held synapse, Bassoon and Piccolo separately or simultaneously share functions in the SV replenishment during high-frequency synaptic activity
[32][41]. In Piccolo-lacking calyxes, the recruitment of slowly releasing SVs in the RRP, that is normally invisible for AP-induced release, is visible during high-frequency stimulation, indicating a role for Piccolo in establishing a sub-pool of the RRP for preventing depletion of release-ready SVs during prolonged and intense firing activity
[32][41]. Additive roles of Piccolo and Bassoon in SV replenishment are revealed in the fast central auditory synapse: Piccolo unlikely influence the release probability, while Bassoon likely regulate it
[33][42].