1. Circulating Neutrophils Increase in Obesity
1. Neutrophils in Obesity
1.1. Circulating Neutrophils Increase in Obesity
Neutrophils are the most abundant leukocytes in human blood, the primary effector cells of acute inflammation and the first responders to infections
[1][2][99,100]. Neutrophils are typically considered to be the major leukocytes against infections due to their capacity to act as phagocytic cells
[3][101], degranulate releasing lytic enzymes, perform an oxidative burst producing reactive oxygen species (ROS), and produce neutrophil extracellular traps (NETs) with antimicrobial potential
[4][5][102,103]. Neutrophils are also considered as the main effector cells of acute inflammatory reactions since they are the first leukocytes to be recruited to inflammation sites where they are capable of producing large quantities of cytokines and chemokines including TNF-α, IL-1β, IL-8, and MCP-1
[6][104]. Consequently, neutrophils induce the second wave of immune cells, such as macrophages and lymphocytes, to inflammation sites
[7][8][105,106].
Conspicuously, circulating neutrophils are increased in obesity
[9][10][11][12][13][107,108,109,110,111], with a clear association between the level of neutrophil blood counts and the higher
body mass index (BMI)BMI [9][10][12][107,108,110]. Furthermore, overweight individuals with neutrophilia presented elevated serum C-reactive protein (CRP) concentrations and larger waist circumferences
[9][10][107,108]. In addition, neutrophil counts were significantly higher in individuals with metabolic syndrome than in lean individuals
[10][108]. In animal models, neutrophils have also been found to be elevated in blood vessels and infiltrating adipose tissue and the endothelium at atherosclerotic lesions
[14][112]. Moreover, neutrophils in obese individuals present an activated phenotype as indicated by elevated plasma concentrations of myeloperoxidase (MPO) and neutrophil elastase (NE)
[15][16][26,113], as well as an increased expression of CD66b, a marker of neutrophil degranulation
[15][17][26,27]. Activation of neutrophils from obese individuals was also indicated by stimulation of the NF-κB signaling pathway
[17][27] and by a higher ROS generation and enhanced release of proinflammatory cytokines
[18][114].
Importantly, weight loss following gastric band surgery resulted in a decrease in neutrophil blood counts
[19][115] and in proinflammatory activities of peripheral blood neutrophils
[18][114]. These results suggest that the inflammatory condition of obesity also leads to the expansion of neutrophils
[20][21][22][9,13,94]. In fact, elevated concentrations of acute-phase proteins have been reported in obese individuals
[23][24][14,116]. As mentioned before, the adipose tissue in obese individuals is capable of producing increased levels of mediators of inflammation like TNF-α, IL-1β, IL-6, and IL-8
[25][26][27][28][29][30][117,118,119,120,121,122]. These inflammatory mediators increase bone marrow granulopoiesis
[31][32][33][123,124,125], releasing neutrophils from the bone marrow to the peripheral circulation. Moreover, these inflammatory mediators induce de-margination of neutrophils from endothelial walls, resulting in neutrophilia
[34][126]. In addition, the adipose tissue also produces leptin which has been shown to promote hematopoiesis
[35][36][37][127,128,129]. Leptin can additionally stimulate the oxidative burst of neutrophils, induce chemotaxis, and inhibit apoptosis in these cells
[35][127]. Together, these reports support the notion that, indeed, obese adipose tissue is responsible for promoting a systemic inflammation that results in the generation, increase in numbers, and activation of neutrophils. In consequence, these leukocytes are the first cells to infiltrate adipose tissues.
2. Neutrophil-to-Lymphocyte Ratio (NLR)
1.2. Neutrophil-to-Lymphocyte Ratio (NLR)
The clear connection between obesity and an elevated neutrophil blood count has motivated people to look for simple biomarkers of obesity and inflammation. The hematological parameter for systemic inflammation known as the neutrophil-to-lymphocyte ratio (NLR) is an easy biomarker of immune response to various infectious and noninfectious stimuli
[38][130]. The NLR is commonly used in many medical areas as an indicator of dynamic changes of neutrophils and lymphocytes in blood during systemic inflammation. The NLR reflects the relationship between innate (neutrophils) and adaptive (lymphocytes) immune responses in various pathological conditions. Because the NLR correlates with CRP concentrations, it becomes a simple cost-effective biomarker for the detection of subclinical inflammation
[39][131].
Accordingly, the NLR has been found to be significantly higher in obese individuals than in healthy lean individuals
[40][41][42][43][132,133,134,135], with a positive correlation to the BMI
[40][43][44][45][46][132,135,136,137,138]. As expected, the NLR is also associated with higher plasma CRP concentrations
[13][40][47][48][111,132,139,140]. The same trend has been reported in mice fed with an obesogenic diet. The effect on the NLR seems to be due to changes in the gut microbiota, which affects blood leukocyte numbers
[42][134]. The significant association between obesity and a high NLR (higher than 4) was a good predictor of increased breast cancer risk. Patients with a high NLR and a high BMI also had the worst disease-free survival
[41][46][133,138]. More importantly, NLR values were found to correlate significantly with the degree of abdominal obesity
[49][50][141,142]. Furthermore, in morbid obese patients, a high NLR was reported to be a powerful and independent predictor of type 2 diabetes mellitus (T2D)
[51][143]. Hence, the NLR is a simple and accessible biomarker that provides information about the inflammatory state of obese individuals. Importantly, the NLR seems to be able to identify an ongoing systemic inflammation in overweight individuals that otherwise appear healthy
[47][48][139,140]. Therefore, a higher NLR in overweight individuals may reflect the subclinical inflammation already present in this group of people.
3. Neutrophil Infiltration into Adipose Tissue
1.3. Neutrophil Infiltration into Adipose Tissue
During obesity-induced inflammation in animal models, neutrophil numbers increase in the peripheral circulation. From there, neutrophils can infiltrate the adipose tissue
[52][25] and blood vessel endothelium
[14][112]. This suggested that neutrophils have an important role at the early stages of obesity by infiltrating the abdominal adipose tissue. Neutrophils are found in the adipose tissue of lean mice in very small numbers, approximately 1% of all immune cells in the adipose tissue
[53][144]. Yet, in mice fed a high-fat diet, a 20-fold increase in adipose tissue neutrophils was observed as early as three days after initiation of the diet
[52][25]. In contrast, macrophage infiltration can be detected after 7 days of a high-fat diet
[54][55][145,146]. These results indicated that neutrophils are the first immune cells to be recruited into adipose tissues. Neutrophil infiltration was first described as transient because after an initial remarkable increase, the neutrophil numbers decreased
[52][25]. However, the neutrophil numbers were higher for up to 12 weeks in the adipose tissue of the mice fed with a high-fat diet than in the adipose tissue of the mice fed a normal diet
[52][25]. Moreover, it was later shown that early recruitment of neutrophils could be prolonged over 90 days with a constant high-fat diet
[56][57][19,147]. Hence, in the early phases of adipocyte disfunction, the initial inflammatory response is characterized by neutrophil infiltration into adipose tissues.
Neutrophils are recruited to the adipose tissue via the action of several chemotactic factors produced in the obese adipose tissue. Inflamed adipocytes produce larger amounts of IL-8, a potent neutrophil chemoattractant
[25][56][19,117]. Once in the adipose tissue, neutrophils can recruit more blood neutrophils by releasing C–X–C motif chemokine ligand 2 (CXCL2), another important neutrophil chemoattractant
[58][148] (
Figure 1). Furthermore, lipids extracted from human adipocytes were shown to induce migration of neutrophils and macrophages, and also secretion of other cytokines
[59][149]. In addition, free fatty acids derived from adipocyte lipolysis could also attract neutrophils and stimulate them to produce more IL-1β, which in turn activates other adipocytes and immune cells
[60][30] (
Figure 1). The exact molecular nature of the various lipid chemotactic factors is not yet known. Future studies will help elucidate these chemotactic factors and the mechanisms they use to recruit neutrophils into adipose tissues.
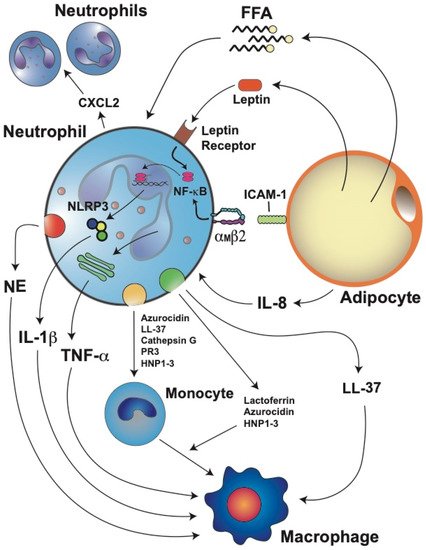
Figure 1. Stressed adipocytes in obese adipose tissue recruit neutrophils, which then further promote inflammation. Adipocytes produce adipokines such as leptin and cytokines such as interleukin-8 (IL-8). IL-8 is a potent chemoattractant for neutrophils. Once in the adipose tissue, neutrophils can recruit more blood neutrophils by releasing C–X–C motif chemokine ligand 2 (CXCL2), another important neutrophil chemoattractant. Neutrophils directly interact with adipocytes via the binding of integrin αMβ2 on the neutrophil to intercellular adhesion molecule 1 (ICAM-1) on the adipocyte. This interaction activates neutrophils and induces them to produce interleukin 1 beta (IL-1β) via the nuclear factor kappa B (NF-κB) and inflammasome (NLRP3) pathways. IL-1β is an important activator of macrophages. Neutrophils also produce tumor necrosis factor alpha (TNF-α), which further stimulates macrophages. Leptin, through its receptor, also activates the NF-κB pathway, resulting in the inhibition of neutrophil apoptosis. Free fatty acids (FFA) derived from adipocyte lipolysis can also attract neutrophils and stimulate them to produce more IL-1β. Neutrophils also produce elastase (NE) which impairs energy expenditure in the adipose tissue and directly activates macrophages. Granule protein cathelicidin (LL-37) can also activate the release of more proinflammatory cytokines from macrophages. Activated neutrophils can also recruit monocytes through the release of azurocidin, LL-37, cathepsin G, proteinase 3 (PR3), and human neutrophil peptides 1–3 (HNP1–3). In addition, lactoferrin, azurocidin, and HNP1–3 can induce polarization of macrophages towards the M1 proinflammatory phenotype.
4. Neutrophil Activation and Inflammation
1.4. Neutrophil Activation and Inflammation
Once in the adipose tissue, neutrophils interact with adipocytes via the binding of integrin α
Mβ2 (Mac-1) on the neutrophil to intercellular adhesion molecule 1 (ICAM-1) on the adipocyte
[52][25] (
Figure 1). This interaction activates neutrophils and induces them to produce IL-1β and TNF-α, which further stimulate inflammation in the adipose tissue
[61][62][63][98,150,151]. Neutrophils also secrete NE, which impairs the energy expenditure in the adipose tissue
[64][152]) and promotes insulin resistance by degrading insulin receptor substrate 1 (IRS-1)
[57][147]. As the number of infiltrated neutrophils augments, the activity of NE is also increased in the adipose tissue of high-fat diet mice
[56][57][19,147]. Importantly, genetic deletion of NE reduces macrophage infiltration into the adipose tissue of obese mice and reverts insulin resistance
[57][65][147,153], indicating that NE is a key activator of macrophages (
Figure 13). An important connection between IL-1β and the NLRP3 inflammasome was found in mice fed a high-fat diet. In the adipose tissue of these mice, the mRNA levels of both IL-1β and NLRP3 were positively correlated to body weight and adiposity
[66][154]. Furthermore, when the mice were fed a calorie-restricted diet, the mRNA levels of both molecules were significantly decreased
[66][154]. Together, these results suggest that the interaction of neutrophils with adipocytes induces IL-1β expression via the NF-κB pathway and that free fatty acids released after lipolysis of adipocytes also stimulate neutrophils to produce high levels of IL-1β via the inflammasome pathway
[60][30] (
Figure 13).
Therefore, the chronic low-grade inflammation of the adipose tissue leads to the activation of neutrophils
[67][17]. Neutrophil activation was first inferred from the observation that serum NE concentrations
[15][26] or plasma MPO concentrations
[68][155] were increased in obese individuals. More recently, these observations were confirmed at the cellular level. In peripheral blood leukocytes, the NE and MPO mRNA levels were found to be positively correlated to the BMI and serum triglyceride concentrations
[16][113]. Furthermore, bariatric surgery, which leads to weight loss in patients, partially reduced neutrophil activation
[15][26]. Another evidence of neutrophil activation in adipose tissues is the fact that leptin can delay apoptosis of mature neutrophils. The antiapoptotic properties of leptin on neutrophils involve activation by the leptin receptor of the NF-κB and MEK1/2 MAPK signaling pathways
[35][69][127,156] (
Figure 1). Activation of neutrophils is also detected by the altered responses neutrophils of obese patients have to various stimuli. In general, these neutrophils display elevated ROS production and release of proinflammatory cytokines
[18][114]. The elevated ROS production observed in neutrophils from obese people has also been reported in neutrophils from obese individuals of other species, including dogs
[70][157] and horses
[71][158].
Another important antimicrobial function of neutrophils is phagocytosis. There are only a handful of reports describing this function in neutrophils from obese individuals. In one study, neutrophils from obese noninsulin-dysregulated horses had a significantly increased ROS production, but no changes were observed in terms of phagocytosis
[71][158]. In another very early study, the phagocytosis and killing of
Candida albicans by neutrophils from healthy (control) and diabetic individuals were compared. Phagocytosis occurred at similar levels in neutrophils from diabetic and control individuals
[72][159]. However, the killing of
Candida by diabetic neutrophils was impaired
[72][159]. In contrast, a recent report of neutrophils from mice on a high-fat diet showed that the phagocytosis of
Klebsiella pneumonia was reduced
[73][160]. In most instances, independently of the level of phagocytosis reported, the killing capacity of neutrophils from obese individuals seems to be diminished. These findings agree with clinical observations consistently reporting that individuals with obesity are physiologically frail and have a higher risk of infections and mortality than normal-weight individuals
[74][75][161,162]. Clearly, much work on neutrophil phagocytosis in obese individuals is needed to fully understand why the activated state of neutrophils from obese individuals does not translate into a more effective antimicrobial function.
Because activated neutrophils have the ability to increase their secretion of cytokines and chemokines, they are also described as the prime effectors of inflammatory responses
[61][98]. As such, they are able to induce the recruitment and activation of the second wave of immune cells, including macrophages, dendritic cells, and lymphocytes
[55][76][146,163]. Once at the inflamed adipose tissue, neutrophils recruit monocytes through the release of LL-37 (cathelicidin/CRAMP), azurocidin (heparin-binding protein), cathepsin G, proteinase 3 (PR3), and human neutrophil peptides 1–3 (HNP1–3)
[77][78][164,165] (
Figure 13). Neutrophils can then induce monocyte differentiation and macrophage polarization and activation. Lactoferrin
[79][166], azurocidin
[80][167], and HNP1–3
[81][168] can induce polarization of macrophages towards the M1 proinflammatory phenotype (
Figure 1). In addition, LL-37 induces M1 macrophage polarization and release of proinflammatory cytokines
[82][169] (
Figure 1). Similarly, alarmin S100A9 also induces the release of proinflammatory cytokines from synovial macrophages
[83][170]. Thus, neutrophil proteins contribute to inflammation intensification by promoting macrophage activation and release of proinflammatory cytokines (
Figure 1).
5. Neutrophil Extracellular Traps (NET)
1.5. Neutrophil Extracellular Traps (NET)
As mentioned, another way neutrophils control infections is the production of neutrophil extracellular traps (NET), which are fibers of decondensed chromatin (DNA) decorated with histones and antimicrobial proteins from neutrophil granules. NET are formed and released by a dynamic cell death program known as NETosis. In addition to infections, NETosis can take place during noninfectious sterile inflammation, where neutrophils help repair damaged tissues. However, during persistent inflammation, NET can aggravate the tissue damage
[84][85][171,172]. Because obesity is associated with chronic systemic inflammation, it is possible that NETosis is activated, and NET may contribute to some of the medical complications associated with obesity.
The role of NET in obesity is not clear since there are conflicting reports. For example, in a diet-induced obesity mouse model, endothelial dysfunction was observed. In these mice, plasma concentrations of LL-37 were increased in mesenteric arterial walls. LL-37 was used as a marker for NET
[86][173]. Disruption of NET with DNase restored the endothelium function, suggesting that NET are increased in obesity and are responsible for endothelial dysfunction
[86][173]. Similarly, in obese persons, plasma concentrations of MPO–DNA complexes (assessed by ELISA) were higher than in lean persons. Moreover, NET concentrations correlated with the BMI
[87][174]. In addition, recent bioinformatics studies found a strong relationship between obesity, inflammatory markers, such as TNF-α, IL-6, IL-8, heat shock protein 90 (HSP90), and NET formation
[88][89][175,176]. Moreover, it was also found that exercise reduces NET
[88][175]. Together, these reports suggest that obesity-induced inflammation is associated with elevated NET formation. However, in other studies, an opposite relationship was reported. Using purified neutrophils from obese individuals and in vitro testing, it was found that although neutrophils displayed an activated phenotype (elevated ROS production), they exhibited lower NET formation than neutrophils from lean individuals
[18][114]. Similarly, using intravital microscopy in mice kept on a high-fat diet, it was revealed that neutrophils produce fewer NET in liver vasculature than neutrophils from lean mice (kept on a normal diet)
[90][177].
Extending these observations to neutrophils from diabetic individuals, it is also found that the role of NET in this obesity-related condition is, again, not clear. Detecting NE–DNA complexes as an indicator of NET, it was reported that recently diagnosed T2D patients had higher plasma levels of NETs than healthy (control) individuals
[91][178]. Furthermore, measuring NE and histone/DNA complexes in serum, it was concluded that NETs were increased in patients with diabetic retinopathy
[92][179]. In another report, neutrophils from diabetic patients with proliferating retinopathy also presented increased NET production, particularly when exposed to high levels of glucose
[93][180]. These results are similar to those reported in a previous study, where diabetic patients had elevated NET components in serum, and neutrophils presented enhanced NETosis in case of high levels (25 mM) of glucose
[94][181]. Together, these reports suggest that diabetic conditions, particularly high glucose, lead to enhanced NETosis. However, other reports indicate that high glucose concentrations decrease the formation of NETs
[95][96][182,183].
Nevertheless, the role for NET in diabetic wound healing seems more clearly established
[97][98][99][184,185,186]. Diabetic patients frequently have foot lesions that do not heal. These individuals are referred to as diabetic foot patients
[100][187]. In a model of wound healing, skin wounds were inflicted on mice. Healing of these lesions was longer in diabetic mice than in normoglycemic (control) mice
[97][184]. Furthermore, when the wounds were treated with DNase 1 to degrade NET, wound healing was improved both in the diabetic mice and the control mice
[97][184]. In addition, the wounds of diabetic animals presented larger amounts of citrullinated histone H3 (H3Cit), a marker for NETosis. In contrast, no H3Cit was observed in the wounds from the
Padi4−/−-mice (deficient in enzyme peptidylarginine deiminase 4 (PAD4) that causes histone citrullination), despite many neutrophils present. More importantly, the wounds of the
Padi4−/−-mice healed very fast
[97][184]. In another study, neutrophils were stimulated with PMA to induce NETosis, and NET formation was abolished by treatment with hydrogen sulfide (H
2S)
[99][186]. Furthermore, diabetic mice with wounds were treated intraperitoneally with H
2S. The wounds in these mice had decreased NET markers (NE, MPO, H3Cit, and PAD4) and healed better than the wounds in the control mice
[98][185]. In a more recent study, intravital microscopy detected enrichment of NET components in the bed of excisional wounds, and inhibition of PAD4 with BB-Cl-amidine improved wound healing in diabetic mice
[99][186]. Together, these results suggested that diabetes slows down wound healing by activating NETosis. Mechanistically, NET slow down wound healing in diabetic animals by triggering NLRP3 inflammasome activation via the TLR-4/TLR-9/NF-κB signaling pathway in macrophages. As a result, macrophages release IL-1β and prolong the inflammatory response in the diabetic wound
[101][102][188,189].
At the present time, it is not possible to decide with confidence whether NET formation is enhanced or reduced in obesity. The discrepancy among the various studies may be related to the different methodological approaches used to evaluate NET. In some studies, indirect assessments were made by detecting some NET components in plasma or serum. In other studies, NET formation was evaluated directly in vitro with purified neutrophils. Yet, in other studies, intravital microscopy was used to detect NET. Methods detecting NET components do not necessarily confirm that NETosis took place. Elevated circulating DNA or neutrophil granule proteins may be caused by several other reasons besides NETosis. In vitro assays with purified neutrophils are more reliable to detect NET. In either case, authors should be aware of the limitations of the methodology used and take them into consideration when interpreting the experimental results. Another possible reason for the discrepancy among the reported results is that neutrophil function may be affected by the metabolic and inflammatory states of the individual. Earlier, it was shown that NET formation is dependent on glucose. Upon PMA stimulation, neutrophils increased glucose uptake and their glycolysis rate (as measured with a Seahorse analyzer). In the absence of glucose, PMA induced neutrophils to decondense chromatin, but they did not release NET. However, if glucose was added at this time, NETs release took place within minutes
[103][190]. Based on these data, the authors suggested that NET formation could be metabolically divided into two phases: the first, independent from exogenous glucose (chromatin decondensation), and the second (NET release), dependent on exogenous glucose and glycolysis
[103][190]. More recently, it was reported that neutrophils from mice fed a normal diet used glycolysis for NET release in both physiological and inflammatory (sepsis) conditions. However, neutrophils from mice fed a high-fat diet could not release NET after a secondary ex vivo activation despite the high glycolytic potential and the flexibility to oxidize fatty acids
[104][191]. Thus, the metabolic and inflammation states of the individual can influence the neutrophil function. In the future, extensive well-controlled studies will be required to elucidate the role of NET in obesity.