1. Introduction
Nematodes are non-segmented roundworms that include a widespread group of metazoans with various habitats ranging from land to water. Nematodes are reported to be the most prevalent and copious in these habitats, with species ranging up to one million
[1]. Nematodes are the least studied organisms despite such a vast distribution. According to studies, less than 0.01% of these species have been reported
[2]. There have been roughly 26,000 species reported so far, out of which 4100 are reported as plant parasites. At the same time, insect-pathogenic nematodes can be used as beneficial organisms in crop production for biological plant protection
[3].
Most soil nematodes contribute significantly to nutrient cycling in the natural environment. Some nematodes are reported to be very important in medical and veterinary sciences
[4]. Precise detection is, thus, critical for understanding nematode biodiversity and developing important management practices. Primary identification and characterization are mainly based on morphological features, including external body structure; total length; differences in sex organs and stoma; organization of lips, mouth, and tail; and other visible morphological characteristics. This often leads to the absence of precise categorization among strongly linked taxa due to specific observable characteristics and a lack of trained taxonomists and thus becomes insufficient, especially when a large sample size is implicated
[5]. Molecular and biochemical identification methods have recently been utilized to overcome the lacuna of morphological identification of nematodes. Other studies have also reported using nematode ribosomal DNA (rDNA) sequencing to evaluate nematode characterization and evolutionary relationships
[6].
Consequently, it is important to correctly identify the nematodes and enhance their evolutionary lineages to understand and combat their adverse effects on animal health and plant yield, in addition to fruitful use in nutrient cycling and other related phenomena.
Caenorhabditis elegans (
C. elegans), a free-living nematode, has proved a good model organism that is preferred by many researchers
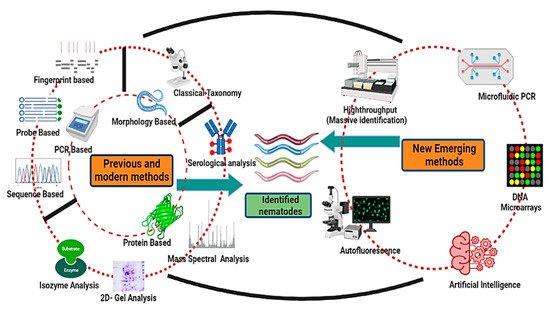
[7]. Because of its simple structure, good heritable traits, the existence of a comprehensive molecular toolset, and an entire genome sequence, it is appropriate for immediate and efficient regulation of gene expression function. The various techniques viz morphological, DNA-based, protein/biochemical-based, and new emerging methods used to identify and classify nematodes are summarized in
Figure 1.
Figure 1.
Different nematode identification techniques (previous, advanced, and emerging).
2. Conventional or Morphometric Method of Identification
Light microscopy has traditionally been used to diagnose and classify nematodes based on morphological and anatomical characteristics. It allows fast and reliable identification of specimens. It is cost-effective and allows a clear distinction between the function and morphological characteristics of the studied specimen
[8].
2.1. Morphological Methods of Identification
Morphological Methods of Identification
Nematode identification methods vary from the detection of a single plant parasite in the soil or from the plants to the whole community observations
[9]. Nowadays, different methods for defining and classifying nematodes are attainable, from basic morphological investigative techniques to much more complex high-performance sequencing methods
[4][10][11][4,10,11]. Nathan Cobb (the father of nematology) has performed a lot of work identifying various free-living and parasitic nematodes, mostly based on their morphological characters
[12]. The significant morphological identification parameters in nematodes include the body length, shape of the head, stylet length, shape of the stoma, number of annules, stylet shape and knob, lateral field structure, shape of the female tail terminus, shape and size of the spicule
[13][14][13,14] and gubernaculum, and spermatheca shape
[15]. Measuring such parameters and processes of nematodes requires trained taxonomists, which is on decline. Morphological parameters can also show variations due to different host plants, nutrition, geographical locations, and other environmental factors as is reported in few free-living and plant parasitic nematodes
[16]. Therefore, it can become difficult for untrained individuals or researchers to identify and characterize a particular nematode species on morphology parameters alone
[17] and thus, an integration of molecular analysis such as DNA sequences can be promising for accurate and proper identification
[18].
3. Molecular Identification Methods
Molecular methods have advanced over classical or conventional taxonomic approaches for the characterization of nematodes
[19]. Polymerase chain reaction (PCR) is widely established and efficiently employed to classify nematodes
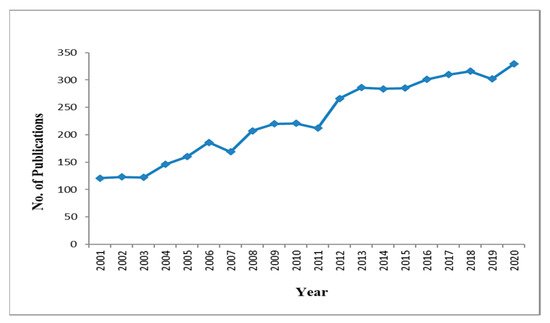
[20][21][20,21]. Good-quality research has been carried out using these molecular markers during the last two decades (
Figure 2).
Figure 2.
Publications on nematode identification as per PUBMED (Query: Molecular markers, DNA barcoding).
3.1. PCR-Based Methods
PCR-based markers are important and reliable sources, permitting the classification and characterization of new species in nematode taxa, such as Rhabditid, Meloidogyne, Pratylenchus, Globodera, and Heterodera
[22][23][24][22,23,24]. DNA-based technologies such as real-time polymerase chain reaction and multiplexed tandem PCR are efficient in the preliminary screening of strongylid nematodes in farm animals, displacing conventional larval culture methodologies. Compared to traditional diagnostic processes, PCR-based detection technologies have revolutionized the field of worm diagnostics
[25]. These are used due to their improved qualities of sensitivity, specificity, speed, relative ease of use, and cost-effectiveness. Using this technique, many copies of a specified DNA template in vitro can be generated. During the last decade of the 20th century, some experiments have suggested the incorporation of effective methods apart from classical methods to decipher the nematode taxonomy
[26][27][26,27]. The data generated through molecular analysis enhances the understanding of nematode systematics and its biology by exhibiting a particular target DNA sequence that will be favorable for the identification of nematode species
[28]. When used to supplement conventional descriptive information, PCR-based molecular methods illustrate the improvements made in this field, providing sensitivity, correctness, and time savings. A significant accomplishment demonstrated that a PCR-based approach effectively elevates polymorphism patches that separate closely related worm species
[29]. Identifying a particular species within a diverse array of nematodes from a soil community is indeed a breakthrough, and was first established using confined ratios of pure DNA and later verified by identifying single individuals from a potent mixture of worms within soil
[30]. In PCR, amplification is performed using nematode DNA or the worm itself as a template
[31]. Several researchers have suggested revised versions of the nematode classification and identification based on the 18S rRNA sequence similarities, which were again focused on using the PCR method
[32]. Based on molecular biology and the need for taxa to be identified, emerging methods such as PCR techniques and sequence-based (ITS and COX), probe-based (qRT-PCR and multiplex PCR), and fingerprint-based (RFLP, AFLP, and RAPD) methods (
Figure 3) have been developed.

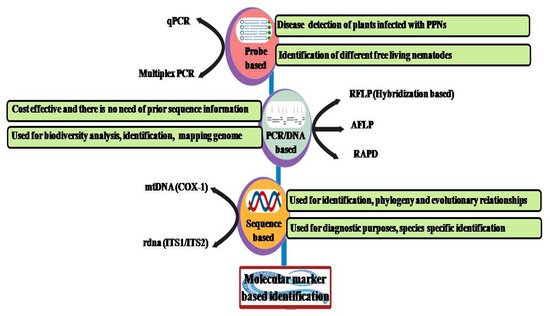
Figure 3.
Different molecular markers for the identification of nematodes based on probe-, DNA-, and sequence-based methods.
3.2. Fingerprint-Based Methods
3.2.1. RFLP (Restriction Fragment Length Polymorphism)
This is one of the first molecular techniques that was used to differentiate different nematode species depending on the usage of other restriction enzymes that help in the digestion of whole genomic DNA or a particular amplified product. This results in the generation of banding patterns depending on the degree of divergence in sequences among different isolates. This method works on the principle of sequence polymorphism, forming definite and distinct cleaving sites for restriction enzymes that produce variable fragments of different sizes
[33]. Many authors used PCR-RFLP to improve Anisakis identification
[34]. This method is simple to use to evaluate the H1 gene and second intergenic spacer of the lungworm Metastrongylus, which provided resistance to
Globodera rostochiensis, a parasite of the potato cyst nematode, and distinguished between three populations of the
Meloidogyn earenaria race while using this technique
[35]. An individual study was carried out on 15 nematode isolates representing 6 species of Trichostronglus. RFLP was used to investigate the diversity of filarial parasite populations that were morphologically indistinguishable. When
Mbo I and
Tag I, two restriction endonucleases, were used in a combinatorial pattern with two probes
viz, rDNA from
C. elegans and pBM103 yielded fragments that were used to discriminate six filial species
[36]. ITS-RFLP is an efficient tool for the classification of different nematodes, as in the case of
Bursaphelenchus, where these nematodes were identified up to the species level. ITS-RFLP is used to discriminate between pathogenic and non-pathogenic isolates of
B. xylophilus. A study was carried out to clarify the phylogeny and molecular discrimination in cereal cyst nematodes (CCNs) using restriction polymorphism of rDNA in different Heterodera and Gotland strain species. After digestion, this experiment differentiated
H. avenae,
H. lapitons,
H. filipjevi, and the Gotland strain with the restriction enzyme (
TaqI)
[34]. Consequently, these reports suggest that RFLP-based characterization is a versatile tool for the examination of nematodes to understand their lineage.
3.2.2. Random Amplified Polymorphic DNA (RAPD)
This technique randomly amplifies genomic DNA in different genome sites, followed by traditional electrophoretic or “lab chip” analysis on any sequence of short DNA fragments/oligonucleotide primers
[14][37][14,37]. It had been used for successful characterization of
Meloidogyne spp. (plant-parasitic nematodes). For instance, two nematode isolates from northern Indiana with the same virulence for resistant soybean lines were separated from the other two southern Indiana isolates with different virulence activity, which led to the development of marker probes using dot blots
[38]. These experiments demonstrated a diagnosis test using RAPD for different isolates of nematodes with some common virulence factors against resistant varieties.
In contrast, the differentiation of two races of devastating plant parasitic nematode
Globodera rostochiensis (Roj and Ro2/3) was carried out using RAPD with similar conditions
[39]. RAPD markers are suitable candidates for differentiating Meloidogyne species using isolated genomic DNA from nematodes, acting as a template
[40][41][40,41]. RAPD characterization of the single female of the Meloidogyne species led to an observation that amplification patterns from a single female root-knot nematode are stable over three successive generations, leading to the assumption of the mitotic parthenogenetic reproductive mode of this nematode
[42]. A shift from RAPD analysis was imperative as low polymorphism, the dominant mode of inheritance, is less ideal for differentiation patterns, and low reliability is the system’s key drawbacks.
3.2.3. Amplified Fragment Length Polymorphism (AFLP)
AFLP is a strong DNA fingerprinting technique for any organism without prior sequence information. The process entails PCR amplification of restriction fragments from complete digestion of genomic DNA, commonly generated using a combination of two restriction enzymes. The AFLP technique was developed to create high-density linkage maps in positional gene cloning and molecular breeding. For instance, Höglund and co-workers employed this technology to uncover genetic differences in parasitic nematodes, including lungworms
[43]. The principle of selective and accurate amplification underpins the AFLP approach, which was developed in response to challenges with endonuclease digestion of genomic DNA and adaptor ligation
[40]. This method investigates gene expression to discover possible parasitic diseases, such as the potato cyst nematode (
Globodera rostochiensis)
[44]. It also identified the tobacco cyst nematode (TCN) complex
[41]. Even though AFLP and RAPD techniques have several similarities, the results of AFLP appear to be much more reliable under strict experimental parameters. In contrast to RAPD-PCR, AFLP is focused on a small quantity of DNA without requiring prior sequence information.
3.3. Probe-Based Detection Methods
Multiplex PCR and quantitative polymerase chain reaction (qPCR) are the two permitted probe-based detection methods of nematode species found in fish populations, such as
Anisakis,
Pseudo terranova,
Hysterothylacium, and
Contracaicum [45][46][45,46]. Multiplex PCR is efficiently applied in various biological and medical studies as it allows simultaneous amplification of many DNA fragments within one reaction
[47]. A maximum of seven specific forward primers were merged with universal reverse primers that anneal to all species of DNA for the ITS region. Because the amplified species-specific primers were of varying sizes, a potential co-infection by multiple species was established
[46]. qPCR is a polymerase chain reaction that incorporates a fluorescent label into every other amplified DNA copy, enabling exact real-time quantitative estimation of the number of multiple crafted copies for every PCR cycle. To identify and quantify
Anisakis spp. and
Pseudo terranova spp. nematodes parasitizing fish-based products, a TaqMan-based qPCR targeting the ITS-1 and 18S rRNA genes was established
[48]. Correspondingly, ITS-2 was used to recognize
A. pegriffi in fish via qPCR. An investigation into the danger of tomato injury was conducted using
Meloidogyne spp. during an experiment
[49]. Li and Co-workers (2014) developed a method for determining Heterodera glycine concentrations in soil samples taken from agricultural fields
[50]. Utilizing this technique, researchers developed a real-time PCR assay to identify
M. hapla in the root galls and surrounding soil. It enabled the differentiation of
M. hapla DNA among 14 other
Meloidogyne spp. except for
M. eminor. The researcher detected DNA extracted from
M. hapla from about 250 mg of soil, containing about one-third of an egg. Similar observations were carried out in other nematode species using the favorable TaqMan qPCR for detection and quantifications
[51].
3.4. Sequence-Based Detection Method
Sequence-based molecular approaches involve the analysis of nucleotide sequences from particular segment(s) of the nuclear DNA, mitochondrial DNA (mtDNA), or whole genome
[52]. The ribosomal DNA (rDNA) and mitochondrial cytochrome C oxidase subunit I (COX1) genes are usually preferred by most studies for diagnostic purposes as these have variable regions that remain conserved. The identification and PCR amplification is easy as both genes have multiple copy numbers in the nematode genome
[53]. The sequence information generated is thus used to decipher the taxa’s phylogeny
[26]. The rDNA consists of conserved coding regions, such as the 28S, 18S, and 5.8S subunits with variable non-coding regions as an internal transcribed spacer (ITS) and external transcribed spacer (ETS) organized as tandem repeats, with intergenic spacers separating the repeating units
[54]. ITS provides the sequence variability in the rDNA, interspersed by the 5.8S coding region in the rDNA cistron into ITS-1 and ITS-2, making the ITS applicable in molecular systematics for the discrimination of closely related species or sibling species
[55]. For
Caenorhabditis spp., diagnosis involves genetic crosses with unknown biological species, which has been characterized using ITS-2 markers alone
[56]. ITS-1 and ITS-2 of nuclear rDNA have consistently been shown to be reliable genetic markers in parasitic nematodes of livestock and have helped to identify different strongylid nematodes, including species of
Haemonchus,
Teladorsagia,
Ostertagia,
Trichostrongylus,
Cooperia,
Nematodirus, and
Bunostomum [57][58][57,58]. When ITS sequences from a variety of Strongylid nematodes were compared, it was shown that ITS-1 (364–522 bp) is usually larger than ITS-2 (215–484 bp). Because of an internal 204 bp region that is repeated twice, the ITS-1 region of
Ostertagia ostertagi and
O. lyrata (801 bp) is longer than that of other Trichostrongylids, including congeners
[59]. The lengths of the ITS-2 sequences of Teladorsagia/Ostertagia species showed no significant differences. Furthermore, investigations suggest that the sequence variation in both ITS-1 and ITS-2 within a species is less (typically less than 1.5%) than the sequence differences among different species, laying the groundwork for strongylid identification and infection diagnosis. ITS-1 and ITS-2 are valuable genetic markers for the development of strongylid nematode diagnostic PCR-based techniques. They are brief (typically less than 800 bp), repetitive, homogenized, and species-specific
[60].
4. Metabarcoding
Barcoding and metabarcoding are complementary approaches and comprehensive solutions to the development of biodiversity analysis. The two procedures are related because they always employ DNA-based accurate detection and have significant advantages due to their unique sequencers and specialized purposes. Metabarcoding has been touted as a ground-breaking tool, displacing standard DNA barcoding
[61]. As DNA barcoding requires the sequencing of one well-curated individual at a time, the metabarcoding process has the potential to make use of the significant gain provided by second-generation sequencing technology, which can create millions of copies in a single run. Metabarcoding requires massive serial sequencing of intricate bulk materials for which exact recognition and curation are impractical. Metabarcoding has aided investigations of tiny eukaryotic cells, either whole populations or individual groups, with marine eukaryotes often being the focus
[62]. It is used with morphometric evaluations, as evidenced in the research findings of estuarine plankton
[63] and nematodes in aquatic habitats
[64][65][64,65].
Meanwhile, molecular-based species identification is a successful approach, with various metabarcoding-based indices produced. Although nematodes have been extensively investigated at the molecular level
[66], their suitability for a biomarker-based molecular approach has received little attention. Complex microscopic examination and metabarcoding studies on microorganisms in soil and marine resources are only carried out locally. Metabarcoding analysis of microbiological metazoan was concentrated on ribosomal RNA (rRNA) loci and the 18S rRNA gene
[67]. The rRNA gene can be conveniently magnified from a wide variety of organisms using only a diverse range of universal PCR primers. Their chronological use in phylogenetic analysis has contributed to sizeable public sequence repositories containing curated benchmark datasets such as SILVA and PR2
[68]. Even though rRNA loci experience highly synchronized evolution inside higher eukaryotes, contrasting rRNA gene replicas can prevail inside individuals, populations, and species
[69]. Polymorphic sequences repeatedly result in loads of molecular operational taxonomic units (MOTUs) being associated amongst each individual; intragenomic rRNA variants can affect alpha and beta diversity calculations in metabarcoding investigations. A new analysis discussed the diagnosis of gastrointestinal nematode parasites, and several PCR methods and DNA microarrays were investigated
[70]. One major problem of eukaryotic metabarcoding studies is how to identify and circumvent the issue of intra-genomic rRNA variation. Bioinformatic tools for copy correction of rRNA metabarcoding datasets have been developed exclusively for prokaryotes since bacterial and archaeal species can also have multiple rRNA gene copies and exhibit some intra-genomic polymorphism
[71].
5. Biochemical- and Protein-Based Methods of Identification
The detection of microbial species using proteome-based matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF/MS) profiling has already reformed diagnostic microbiology. The molecular weight of proteins such as ribosomal and other abundant proteins is used to identify species. DNA-based approaches, mass-to-charge ratios, protein sequences, and immunological approaches highlight the utilization of different protein contents and shapes to characterize nematode species. Furthermore, protein structure and post-translational alterations extend the spectrum of nematode species that may be identified and make identification easier
[72][84].
5.1. Analysis Based on Isozymes
Isozyme analysis is one of the early approaches employed to identify nematodes that did not rely on appearance. This approach involves the removal of soluble proteins from whole nematodes in buffer solutions, then running the extracts over a starch or polyacrylamide gel, and labeling certain enzymes. The electrophoretic technique, also called multi-locus enzyme electrophoresis, is based on isozyme migration patterns generated by the electrical load, molecular weight, and conformance due to changes in the amino acid composition. Esterases were the most often utilized enzymes, although additional enzymes such as malate dehydrogenase, superoxide dismutase, and glutamate-oxaloacetate transaminase were also used to various degrees. This strategy aided in improving and clarifying evolutionary relationships, notably among economically important Meloidogyne species. Despite this, the procedure was inefficient and time-consuming, with one of its shortcomings being the requirement to compare known samples
[72][84]
5.2. Use of Two-Dimensional Gel (2-DGE) Analysis
Two-dimensional electrophoresis, also called 2-DGE, has been useful in nematode taxonomic studies. The approach allows for charge-based resolution of complex protein mixtures, followed by mass-based characterization in a dimension perpendicular to the first one. The resolution spectrum is then assessed between samples to check whether similarities or variations may be categorized as existing or absent for phylogenic or cladistic analysis of such a resultant data matrix. For instance, Navas and Co-workers (2002) employed 2-DGE to compare the proteomes of 18 root-knot nematodes from 4 strains
[73][76]. According to the researchers, it was sometimes difficult to analyze spots. It was difficult to discern which reported alterations were fundamental due to gel anomalies and other issues. As a result, only 95 spots per nematode were scored, as revealed by the 2 replicates employed per nematode. The technique has advantages and downsides
[73][76]; hence, 2-DGE allows for making evolutionary conclusions about the taxa.
5.3. Serological Analysis or Use of Antibodies
Immunoassays have great application in agriculture for the diagnosis of crop diseases, pesticides, and other naturally occurring compounds. Bird’s first reported generation of antibodies against nematodes was shown in 1964
[74][85]. After that, several researchers have demonstrated promising outcomes using poly and monoclonal antibodies (mAbs). Meloidogyne, Heterodera, and Globodera genera have been classified using this approach. Lee (1965) attempted serological discrimination of Meloidogyne species
[75][86]. Bird reported the probability of developing antiserum against nematodes. Various researchers have investigated the use of polyclonal and monoclonal antibodies against nematodes since Bird initially reported on the possibility of generating antiserum against nematodes with variable results. Lee observed that anti-serum generated against
M. incognita did not form a typical arc-shaped precipitation band when coupled with antigens from another species within the same genus, with
M. hapla demonstrating a lack of cross-reactivity in the Ouchterlony double diffusion assay
[76][87]. The apparent specialty could thus be attributed to a small number of nematode samples. The cyst nematode Heterodera and Globodera species have shown similarly mixed results
[77][88]. The development of Kohler and Milstein’s hybridoma technique raised expectations within the nematology community that mAbs could be developed for medical diagnostics
[78][89]. The method involved fusing matured B cells from animals immunized with nematode antigens with mouse lymphoid tumor cells in vitro to form indefinite hybridomas for a continuous antibody yield. Using the hybridoma technique, immunoglobulins against many significant agronomic nematodes, such as
Meloidogyne incognita,
Globodera rostochiensis,
Heterodera glycines, and
Globodera pallid, have indeed been formed. Schots and colleagues discovered that only specific mAbs distinguished between
G. rostochiensis and
G. pallid isolates
[79][90]. The hybridoma technique becomes more challenging as the number of nematode samples increases. Because the restructure of antigen-site-binding sequences for comparative observations is feasible through the single B cell sequence receptor (ScBCR-Secq), this line of nematode detection algorithms can be reconfigured using next-generation advanced technologies
[80][91].
5.4. Use of Mass Spectrometry Analysis
Pathogenic nematode protein profiles can help researchers to understand how pathogenic nematodes interact with their hosts. Millares and co-workers (2012) classified proteins linked with antibiotic resistance in
Haemonchus contortus, utilizing mass spectrometry and MALDI-ToF
[81][92]. Without having access to a nematode’s whole genome sequence, the MALDI-ToF technology can be used to classify them by investigating their proteomes. Although MALDI-ToF generates minor fragments, the ions generated are often single-charged and non-fragmented, meaning parental ion masses can be easily determined from mass spectra
[82][93].
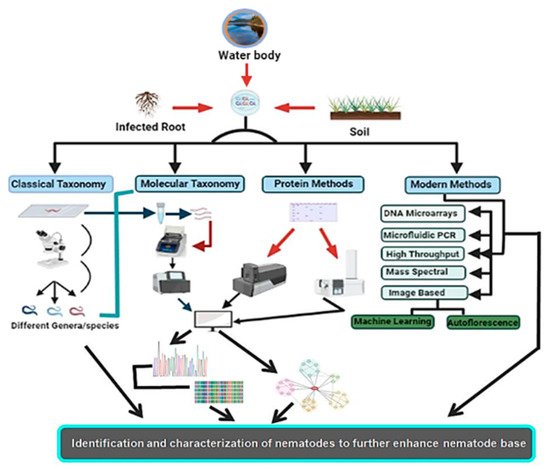
Figure 4 shows the basic setup required for nematode characterization and identification using proteins as markers, with some modern and day-to-day techniques followed for nematode identification.
Anguina tritici,
A. funesta, and
M. funesta were separated using complete second-stage juveniles and/or proteins generated from these in a range of organic solvents
[83][94]. These factors revealed that even a single
M. incognita nematode (adult female or J2), cleaned or unclean, squashed or undamaged, may be utilized for MALDI-ToF-MS treatment investigations
[84][95]. Juvenile and J2s had different protein profiles, with everyone possessing their diagnostic peak; larger mass and higher peaks were found when shattered and/or washed samples were employed. However, leading to a shortage of appropriate information in the database systems at the time, their efforts to categorize proteins using resemblance matches completely failed
[85][96]. 2-DGE and MALDI-ToF-MS were used to examine the proteomes of two neuromorphic species,
Paragordius tricuspidate and
Spinochordodes tellinii.
Figure 4.
Different methods with their associated experimental setup, with the main focus on protein and modern approaches to decipher nematode identity and characterization.
In contrast, the species of hairworm shared 36.2% of the total protein spots in the 2-DGE analyses, where 38.0% were unique to
P. tricuspidate and 25.8% to
S. tellinii; a hereditary distance of 0.47 created 2 different species, verifying the strained relationship originally recorded for such species
[86][97]. They have been engaged in the recognition of MS fingerprints of proteins obtained from digested gel plugs by conducting preliminary database searches.
Protein coding sequences in nematodes differ depending on their genetic makeup and environment. Furthermore, various variables can impact the results, the protein extraction procedure, the effectiveness of the 2-DGE activity, and the instrument design. These studies demonstrated that 2-DGE in conjunction with MALDI-ToF-MS is a dependable roundworm taxonomic classification method.
6. Emerging Methods of Nematode Identification
With the growing demand and utility of nematodes (free-living nematodes) as a model for different studies, which include disease detection, aging, and other important physiological and developmental studies, the need to characterize nematodes with modern methods (
Figure 4) has emerged, which could further enhance the already established base of nematode characterization. In 2006, Gasser and co-workers acknowledged that the genomic, genetic, and protein fields were recognized to define technological advances that provide several applications in veterinary parasitology research. Universal microarrays, nanotechnology systems, and MS frameworks have recently been established for about a decade. They are increasingly being employed in several studies, including parasitology and identification. These technologies could help develop or diagnose completely integrated methods
[11][87][88][11,98,99]. These approaches could impact budding protozoan parasite research. However, by selecting or designing the latest technology, the biological consequence should be appropriately examined
[88][99]. This technology is less sophisticated, complex, and cost-effective than HTS for a small number of samples. The Sanger worm classification or characterization method has sequenced PCR results for over 20 years. A practical method of reading DNA sequences was devised using molecular evolutionary biology and comprehensive analysis of a specific individual. Scientists utilized sequencing to distinguish among distinct harmonic populations and validate the development of a novel
Phaphid ascaris species in marine fishes
[89][100]. Ristau and colleagues used sequencing and phylogenetic analysis to describe the first strongly linked sister species of the freshwater nematode
Tobrilus gracility [90][101]. Nowadays, this technique is precious since it enables the identification of new species or populations without prior information. The Sanger technique might be used to verify next-generation sequencing (NGS). It is easy to analyze because it requires essential bioinformatics and, therefore, can manage a short sequence. A Sanger sequence’s average duration is more significant than a second-generation sequence’s average duration. However, this technique suffers from a critical bottleneck in large-scale detections
[91][102]. The count of nematode species with sequenced and annotated genomes is increasing. This will aid researchers in understanding worms. This will most likely add research into the nematode genome evolution, comparative nematode genomics, and the biology of different parasitic nematodes. It is also helpful in the creation of new molecular markers for these species and population identification and characterization.