Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Fujie Zhao and Version 2 by Camila Xu.
Autophagy is a highly conserved process in which obsolete and dysfunctional cytoplasmic components (such as unfolded proteins, lipids, and damaged organelles) are degraded and recycled, and infectious organisms are removed by lysosomes. Deficient or uncontrolled activation of endothelial autophagy is associated with the onset and development of diverse cardiovascular diseases (CVDs), including coronary microvascular dysfunction” (CMD).
- coronary microvascular dysfunction
- endothelial cell
- autophagy
1. Brief Overview of Autophagy
Autophagy is a highly conserved process in which obsolete and dysfunctional cytoplasmic components (such as unfolded proteins, lipids, and damaged organelles) are degraded and recycled, and infectious organisms are removed by lysosomes [1][2][3][4][10,11,38,39]. Autophagy is stimulated by different stresses (i.e., nutrient deprivation and hypoxia) and functions primarily as a cell survival mechanism [5][6][7][8][40,41,42,43]. However, it switches to promoting cell death under insurmountable lethal stress, which is known as autophagic (type II) cell death [9][10][44,45]. Three classes of autophagy are identified as follows: chaperone-required autophagy, microautophagy, and macroautophagy [11][12][13][46,47,48]. Chaperone-required autophagy is the selective degradation of proteins with a KFERQ-like motif. During the process, targeted proteins are transferred to the lysosomes with the company of the chaperone HSC70 and co-chaperones, subsequently internalized into the lysosomes via an interaction with the lysosome-associated membrane protein type 2A (LAMP2A) [14][49]. In microautophagy, the cargo, alone or in a complex with chaperones, can be directly engulfed by the lysosome and late endosomes through invagination at the lysosomal membrane via electrostatic forces [15][16][17][50,51,52]. Macroautophagy (hereafter referred to as autophagy) is the most well studied and the major type of autophagy. The macroautophagy pathway is characterized by the formation of autophagosomes, within which cytoplasmic components are insulated and subsequently degraded by fusing with the lysosomes [10][18][19][20][12,45,53,54]. The process of autophagy can be dissected into the following sequential steps: induction of a phagophore assembly site, nucleation of an autophagosome precursor (known as the phagophore), membrane expansion and maturation of the autophagosome (a double membrane vesicle), fusion with the lysosome for degradation, and lastly, recycling the degraded cargo [1][2][21][22][10,11,55,56].
The process of autophagy is sequentially regulated by multiple mechanisms (Figure 12). (1) First, initiation of the phagophore assembly begins with the formation of a preinitiation complex, which is composed of the unc-51-like kinase (ULK1/2), autophagy-related protein 13 (ATG13), and the non-catalytic focal adhesion kinase-family interacting protein of 200 kD (FIP200) [10][23][24][45,57,58]. The activity of this kinase complex is negatively inhibited by the mammalian target of the rapamycin complex 1 (mTORC1) and the positively activated AMP-activated protein kinase (AMPK) [25][26][59,60]. (2) Further nucleation involves the recruitment and activation of the initiation complex, which is composed of the vacuolar protein sorting protein 15 (VPS15), a class III PI3K (VPS34), and Beclin 1 [27][28][61,62]. The activity of the initiation complex is downregulated by several independent signaling pathways, such as the PI3K-AKT pathway, and the Bcl2 and Bcl-xL pathway [29][30][63,64]. Conversely, starvation or exercise-induced autophagy activation is carried out by the JNK family [31][32][65,66]. (3) Next, the phagophore elongation and closure require two distinct but complementary ubiquitin-like protein conjugation systems—the ATG12/ATG5/ATG16L1 complex and the microtubule-associated protein 1-light chain 3 (LC3)-phosphatidylethanolamine (PE) machinery [10][45]. (4) Finally, the fusion of the autophagosomes with the lysosomes requires the syntaxin17 (Stx17) synaptosome-associated protein 29 (SNAP29), the vesicle-associated membrane protein 8 (VAMP8), and the lysosomal-associated membrane protein 1/2 (LAMP1/2) [33][34][35][36][67,68,69,70].
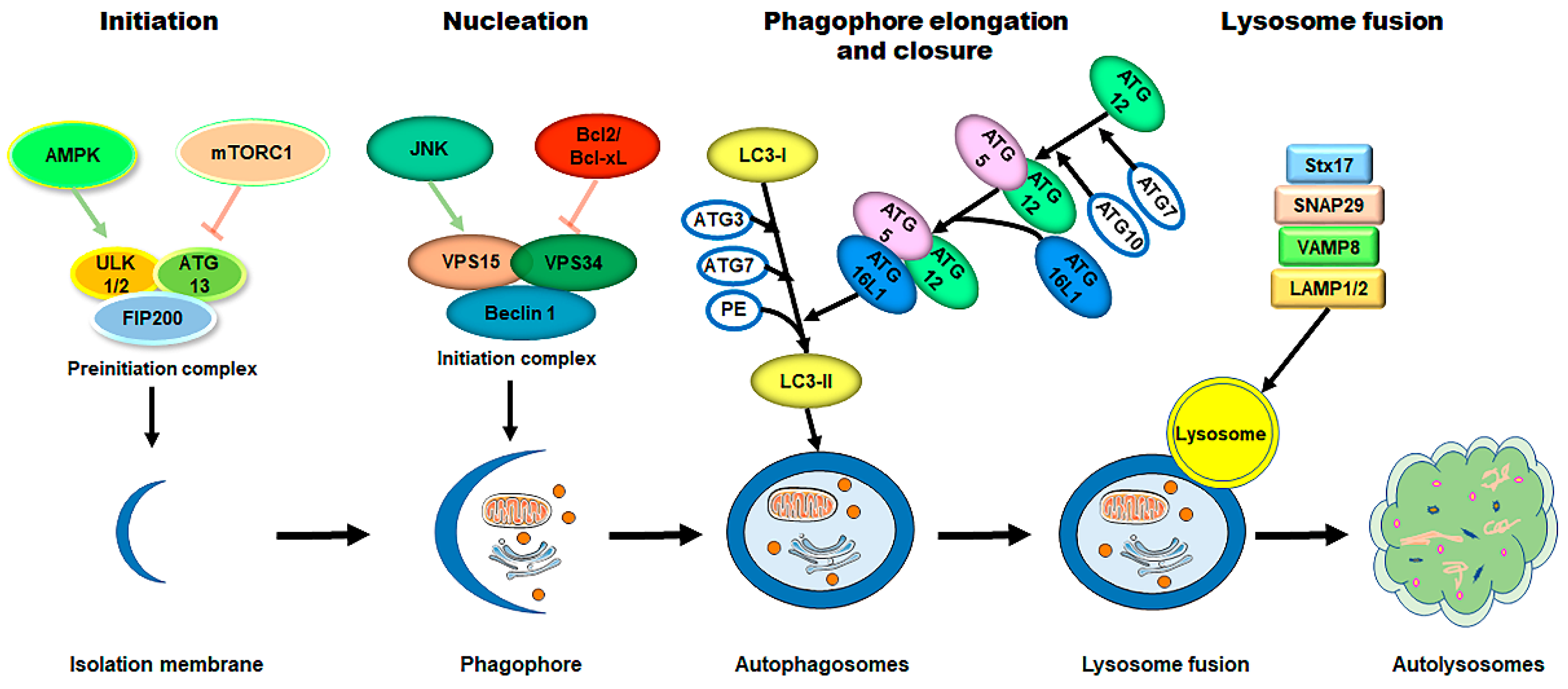
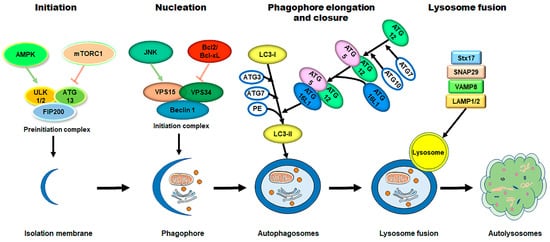
Figure 12. The process of autophagy in mammalian cells. The process of autophagy is sequentially dissected into several phases, including initiation, nucleation, phagophore elongation/closure, and autophagosome–lysosome fusion. (1) First, upon metabolic insults, the AMPK activation and/or mTORC1 inhibition result in the initiation of the preinitiation complex (ULK1/2, ATG13, and FIP200). (2) Further nucleation involves the recruiting and activating of the initiation complex (VPS15, VPS34, and Beclin 1), which is downregulated by the Bcl2/Bcl-xL pathways and upregulated by the JNK family. (3) Next, phagophore elongation and closure require the ATG12/ATG5/ATG16L1 complex and the LC3-PE machinery. (4) Finally, the fusion of the autophagosomes with the lysosomes requires Stx17, SNAP29, VAMP8, and LAMP1/2. Abbreviations: AMPK, AMP-activated protein kinase; mTORC1, mammalian target of rapamycin complex 1; ULK1/2, unc-51-like kinase 1/2; ATG, autophagy-related protein; FIP200, the non-catalytic focal adhesion kinase-family interacting protein of 200 kD; VPS, vacuolar protein sorting; LC3, microtubule-associated protein 1-light chain 3; PE, phosphatidylethanolamine; Stx17, syntaxin 17; SNAP29, synaptosome-associated protein 29; VAMP8, vesicle-associated membrane protein 8; LAMP1/2, lysosomal-associated membrane protein 1/2.
The selective sequestration of the dysfunctional mitochondria by the autophagosomes and the subsequent degradation by the lysosomes are termed "mitophagy” [18][12] (Figure 23). There are two distinct signaling pathways for mitophagy, which are as follows: the phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)–Parkin-dependent pathway and the mitophagy receptor-required pathway [37][38][71,72]. To date, multiple mitophagy receptors have been recognized in mammals, including the BCL2/adenovirus E1B 19kDa-interacting protein 3 (BNIP3) [39][73], NIP3-like protein X (NIX, also called the BNIP3-like protein (BNIP3L)) [40][41][42][74,75,76], FUN14 domain-containing protein 1 (FUNDC1) [43][77], B-cell lymphoma-2-like 13 (BCL2L13) [44][78], and FK506-binding protein 8 (FKBP8) [45][79].
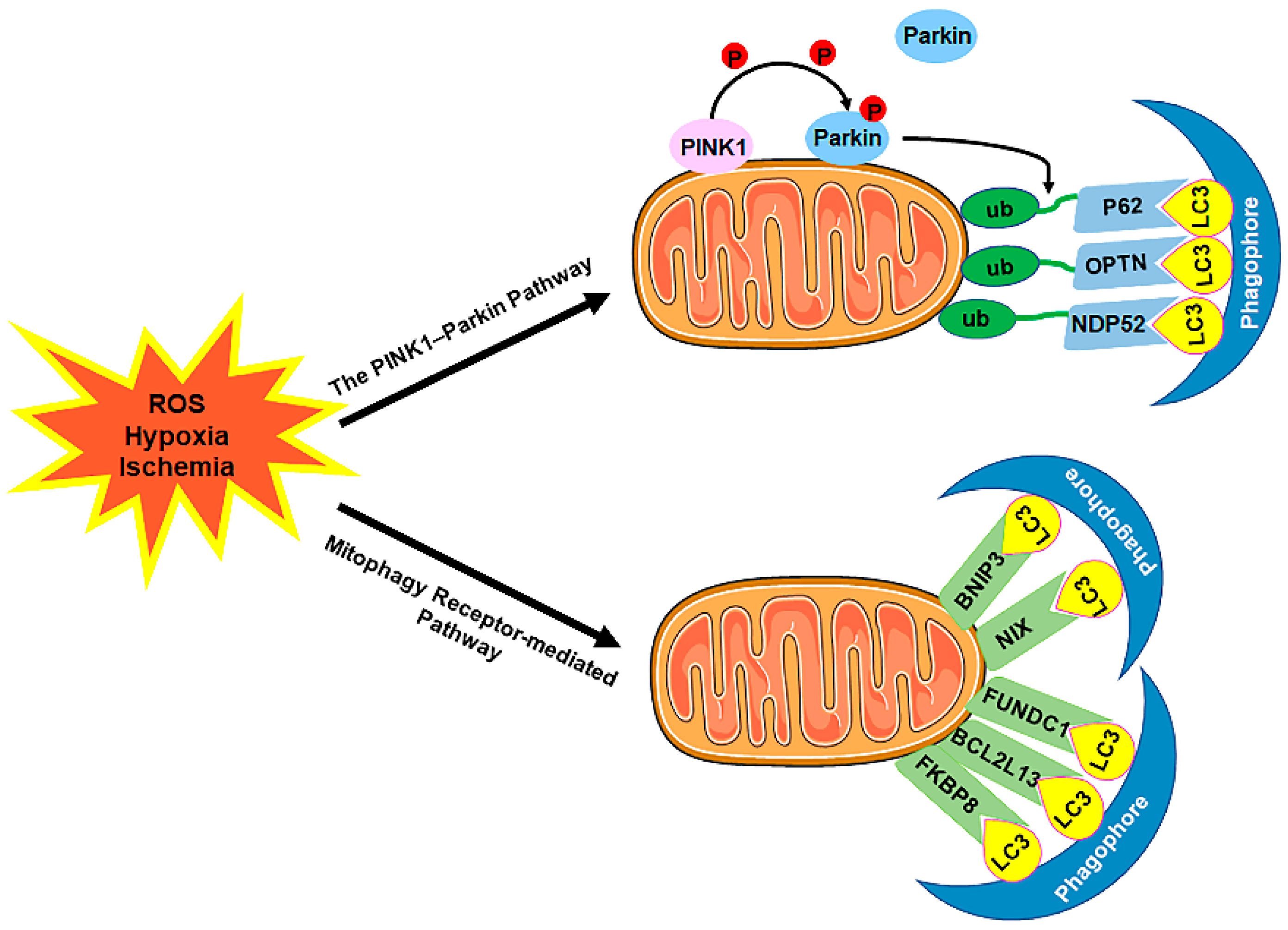
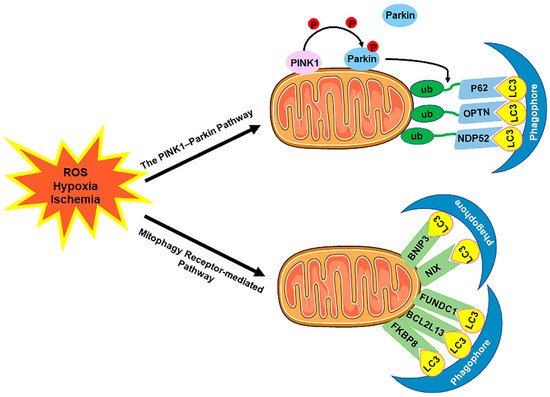
Figure 23. The mechanism of mitophagy. Mitophagy is induced by ROS, hypoxia, ischemia, and other stimuli. There are two distinct signaling pathways for mitophagy, which are as follows: (1) The PINK1–Parkin pathway of mitophagy. Following stress, PINK1 accumulates on the outer membrane of the mitochondria (OMM), promoting Parkin recruitment to ubiquitinate several OMM components. Poly-Ub chains are subsequently recognized by adaptor proteins (p62, OPTN, and NDP52) and further initiate autophagosome formations through binding with LC3. (2) The mitophagy receptor-mediated pathway. The BNIP3, NIX, FUNDC1, BCL2L13, and FKBP8 mitophagy receptors localize to the OMM and directly bind with LC3 to mediate mitochondrial elimination. Abbreviations: PINK1, PTEN-induced putative kinase 1; Parkin, Parkin RBR E3 ubiquitin-protein ligase; ub, ubiquitination; p62/SQSTM1, sequestosome 1; OPTN, optineurin; NDP52/CALCOCO2, calcium binding and coiled-coil domain 2; LC3, microtubule-associated protein 1A/1B-light chain 3; BNIP3, BCL2/adenovirus E1B 19 kDa-interacting protein 3; NIX, NIP3-like protein X; FUNDC1, FUN14 domain-containing 1; BCL2L13, B-cell lymphoma-2-like 13; FKBP8, FK506-binding protein 8.
2. Coronary Endothelial Autophagy in CVDs
In the past, autophagy (including mitophagy) in cardiomyocytes was thought to play a predominant role in heart injuries. With the increasing recognition of the crucial contributions of CMD to CVDs, the role of autophagy (including mitophagy) in non-myocytes, particularly in coronary ECs, has attracted great interest. Emerging evidence unravels that autophagy is required for multiple EC functions, such as secretion of adhesion molecules [46][47][80,81], EC nitric oxide synthase (eNOS)-derived NO bioavailability [48][49][50][82,83,84], expression of ET-1 [51][52][85,86], ROS production [49][53][83,87], and inflammatory cytokines production [49][83], which participate in a wide range of cellular events, including endothelial proliferation [52][86], senescence [53][54][55][87,88,89], and apoptosis [54][56][57][58][88,90,91,92]. EC-intrinsic autophagy is suggested to enable ECs to adjust plastically to various insulting stressors [51][56][59][85,90,93] and leads to autophagic cell death in severely damaged ECs [60][61][62][94,95,96].
2.1. Coronary Endothelial Autophagy in Obstructive CAD (Stable CAD and Acute Coronary Syndromes)
In the hearts of obstructive CAD patients, CMD probably coexists and plays a role in causing myocardial ischemia in regions perfused by arteries both with and without stenosis. Thus, CMD has important diagnostic, prognostic, and management implications [63][64][65][66][1,18,97,98]. In regions distal to arterial stenosis, the chronic modulation of coronary microcirculation to limited perfusion pressure may negatively impact the microvascular remodeling and the maximal capacity of vasodilation after restoring to normal CBF [67][99]. The most severe form of CMD is microvascular obstruction (MVO), which refers to a capillary destruction with a no-reflow phenomenon, despite recanalization of the epicardial coronary artery [68][69][70][100,101,102]. The pathogenic mechanisms underlying CMD in obstructive CAD include coronary microvascular EC injuries, which may occur much earlier and with much more severe damage than cardiomyocyte injuries. Emerging evidence shows that autophagy plays a fundamental role in this process [71][72][103,104]. However, the underlying mechanisms remain controversial. Upon oxidative stress, Wang et al. reported that microRNA-103 could protect the human coronary artery ECs (HCAECs) against H2O2-induced injuries by preventing the Bcl-2/BNIP3-mediated suppression of end-stage autophagy [73][105]. Furthermore, by using mice with EC-specific NADPH oxidase 2 (Nox2)/gp91 overexpression, Shafique et al. demonstrated that endogenous ROS oxidative stress protected mouse heart ECs from oxidant-induced cell death by increasing the AMPK-mTOR-mediated autophagy and through improving the AMPK-eNOS-mediated EC-dependent-coronary vasodilatation [74][106]. Autophagy can also be induced by hypoxia stress [75][107]. Wang et al. identified that forkhead box O3 alpha (foxO3α)-dependent autophagy aggravated hypoxia-induced rat cardiac microvascular endothelial cell (CMEC) dysfunction and apoptosis [76][108]. However, according to Sun’s study, the mitophagy induced by the Rcan1-1L (regulator of calcineurin 1-1L) overexpression contributed to cell survival under hypoxic conditions [77][109].
Notably, the role of endothelial autophagy in hypoxia/reoxygenation (H/R) or ischemia/reperfusion (I/R)-induced injury is much more complex. Generally, autophagy is a protective mechanism against cardiac H/R or I/R injury [78][112]. Its protective role is driven by inducing the mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway [79][113] or via activating and promoting the transcription factor EB (TFEB) translocating from the lysosomes to the nuclei [80][114]. Furthermore, mitophagy was usually referred to as a pro-survival regulator to I/R or H/R injury [81][115]. Inhibition of the FUNDC1-mediated mitophagy in CMECs by the nuclear receptor subfamily 4 group A member 1 (NR4A1) [82][116] or receptor-interacting protein kinase 3 (Ripk3) [83][117] exhibited the disturbed mitochondrial homeostasis, upregulated the expression of EC-derived pro-inflammatory and adhesive factors, enhanced endothelial apoptosis, and provoked CMD in cardiac I/R injuries. In addition, enhancing autophagy through Beclin1 overexpression in CMECs exhibited suppressed NACHT, LRR, and PYD domain-containing protein 3 (NLRP3) inflammasome activation by promoting tumor necrosis factor-alpha-induced protein 3 (TNFAIP3) [84][118] and inhibited caspase-4 inflammasome activation [85][119]. Thus, it resulted in a reduced IL-1β level and an increased animal survival upon myocardial I/R injuries. In addition, the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) overexpression [86][120] and miR-92a-3p inhibition [87][121] could protect the CMECs against myocardial I/R injuries by preserving the EC mitophagy. However, hyperautophagy is linked to I/R or H/R injury-induced mitochondria and EC apoptosis. The inhibition of autophagy contributed to the anti-apoptosis effects of glycyrrhizic acid (GA) in H/R-induced CMEC injury [88][122]. Melatonin was reported to play a beneficial role in CMECs against I/R injury through directly suppressing autophagy via the AMPK/mTOR pathway [89][123] or by inhibiting the mitophagy-mediated cell death via the dynamin-related protein 1 (Drp1)-voltage-dependent anion channel 1 (VDAC1)-hexokinase 2 (HK2)-mitochondrial permeability transition pore (mPTP)-PINK1/Parkin axis in an AMPKα-dependent manner [90][124]. Furthermore, neuregulin-1β (Nrg1β) protected the cardiac ECs against I/R injury by preventing ATG5-required autophagy-induced Trx2 (thioredoxin) degradation and rescuing eNOS function via upregulating the Erb-B2 receptor tyrosine kinase 2 (ErbB2) [91][125].
2.2. Coronary Endothelial Autophagy in HFpEF
HFpEF is recognized as a clinically heterogeneous syndrome, in which patients present classic symptoms and signs of HF but exhibit a normal or near-normal EF [92][93][134,135]. CMD is hypothesized to play a fundamental role in the pathophysiological process of HFpEF [94][95][96][97][136,137,138,139]. Up to 75% of HFpEF patients exhibit impaired CFR in spite of the absence of obstructive CAD [98][140]. Diabetes mellitus (DM), metabolic syndrome, hypertension, and obesity are prevalent cardiovascular risk factors for HFpEF [92][94][99][134,136,141]; they aggravate cardiac dysfunction and remodeling through CMD [100][142]. The basic mechanisms that mediate the progression of HFpEF include myocyte hypertrophy, energetic imbalance, mitochondrial dysfunction, EC dysfunction, increased oxidative stress, inflammation, interstitial fibrosis, and damaged angiogenesis [101][102][103][104][143,144,145,146]. The pathophysiological role of autophagy in HFpEF onset and progression has just begun to be acknowledged. Here, reswearchers mainly focus on the impact of autophagy on cardiac angiogenesis and fibrosis, two critical events associated with the progression of HFpEF.
2.3. Coronary Endothelial Autophagy in DCM
Clinical studies have shown that CMD is an early feature of DCM (even in patients without obstructive CVDs) [105][106][107][171,172,173] and this impairment is more pronounced in type 2 DM patients [108][109][110][111][174,175,176,177]. DCM studies in both animals and humans have emphasized the substantial role of the coronary ECs, particularly in the early stages of damage, in promoting ROS generation, and facilitating the recruitment of inflammatory cells. This ultimately resulted in myocardial microvascular rarefaction, diminished angiogenesis, and HFpEF phenotype [112][113][114][178,179,180]. Insulin signaling impairment, hyperglycemia/glucotoxicity, and lipotoxicity are predominant pathophysiological causes of DM-related CMD. Dysregulated autophagy is a key underlying cause in the onset and progression of DCM. A prolonged exposure of a fetal mouse heart to sugars (sucrose or mannitol) could induce severe lysosomal derangements and prominent autophagy in the ECs [115][181]. Mst1 (mammalian sterile 20-like kinase 1) is a serine/threonine kinase that functions as a negative regulator of autophagy in the heart by enhancing the binding of Beclin1 to Bcl-2 and promoting apoptosis by releasing Bcl-2 from Bax [116][117][182,183]. Hu et al. showed that Mst1-enriched exosomes excreted by CMECs were taken up by cardiomyocytes, resulting in inhibited autophagy and ultimately exacerbated high glucose (HG)-induced apoptosis in cardiomyocytes [118][184]. Meanwhile, Mst1 directly participated in the pathogenesis of CMD by inhibiting autophagy and increasing apoptosis in CMECs [119][185]. Furthermore, the upregulation of autophagy was reported to rescue HG-induced EC apoptosis through the AKT-mTOR signal pathway [120][186]. In addition, mitophagy was shown to protect mitochondrial integrity and prevent HG and palmitate acid (HG/PA)-induced EC apoptosis via the PINK1–Parkin pathway [121][122][187,188] and hinder HG/PA-induced EC senescence via the AMPK pathway [123][189]. Furthermore, improving Bnip3-dependent mitophagy could rescue ox-LDL-induced EC damage, resulting in a restored mitochondrial respiration complex activation, reduced ROS production, and an increased EC viability [124][190]. Interestingly, in certain conditions, the inhibition of autophagy can be protective. The downregulation of autophagy was reported to relieve HG-induced endothelial impairment via the glioma-associated oncogene homolog 1 (GLI1)-dependent-Hedgehog pathway [125][191].
2.4. Coronary Endothelial Autophagy in Other Heart Diseases
Except for the diseases mentioned above, coronary autophagy was also reported to participate in many other diseases. Kawasaki’s disease (KD) is a systemic febrile vasculitis and can lead to abnormalities of the coronary artery in about 25% of untreated cases, which has been reported as the predominant cause of children’s acquired heart diseases [126][127][195,196]. According to Qin’s report, the peripheral blood mononuclear cells (PBMCs) collected from KD patients with fever could induce autophagy in HCAECs, thus, promoting the secretion of chemokines and pro-inflammatory factors [128][197]. Moreover, ginsenoside Rb1 could effectively alleviate coronary artery lesions in a mouse KD model, possibly by upregulating the AMPK/mTOR/P70S6 pathway-mediated autophagy to prevent EC injury [129][198]. Additionally, the activation of autophagy was also involved in the anti-inflammatory effects of resveratrol in TNF-α-treated HCAECs [130][199]. Furthermore, autophagy was found to be upregulated during zebrafish heart regeneration and was positively correlated with the metformin-mediated cardiac regeneration acceleration in zebrafish, including epicardial, endocardial, and vascular endothelial regeneration [131][200]. Moreover, recent studies show that in aged EC compartments, autophagic activities are compromised [55][89]. Accordingly, in comparison with ECs in younger mice, ECs from older mice displayed lower levels of vital proautophagic proteins, such as Beclin1 and LC3 [132][201].
Notably, after coronary angiography, up to 40% of patients with typical clinical manifestations of myocardial ischemia were found with normal or near-normal appearing coronary arteries [133][134][135][136][137][138][202,203,204,205,206,207]. This situation is termed “MVA”, in which CMD is the principal alteration causing symptoms [133][202]. In particular, MVA is the disease that fosters the concept of CMD and draws people’s attention to the role of CMD in nonobstructive heart diseases for the first time. Unfortunately, to date, there is no animal model for MVA. Mechanistic research is urgently needed.