1. The Immunosuppressive Tumor Microenvironment and Physical Barriers Favorable to Resilient Tumor Growth
Circulating
chimeric antigen receptor (CAR
) T cells must gain sufficient traction and adhesion against receptors on the endothelial wall to initiate extravasation (
Figure 1). However, secretion of angiogenic factors VEGF and bFGF during tumorigenesis leads to insufficient expression of adhesion molecules—such as intercellular adhesion molecule 1 (ICAM-1), 2 (ICAM-2), and vascular cell adhesion molecule 1 (VCAM-1)—on endothelial cells which prevents efficient T cell engagement
[1][14]. Adhesion molecules expressed on both immune cells and endothelial walls modulate interactions necessary for transendothelial migration (TEM). Although mechanisms of TEM have been reported extensively, identification of a comprehensible approach to enhance the extravasation of T cells remains elusive. Instead, T cells have been engineered to target tumor-associated endothelial receptors such as VEGFR2 to disrupt tumor-supporting vasculature, enhance T cell trafficking, and deprive the tumor of nutrient and oxygen supplies
[2][3][15,16]. This approach revealed the efficient killing of in vitro tumor spheroids
[3][16] and prolonged survival in animal models
[2][4][5][15,17,18].
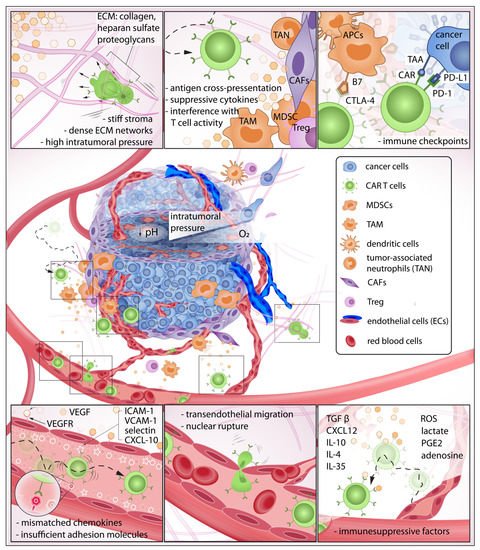
Figure 1. The exhausting journey of CAR T cells in the tumor microenvironment (TME): In this journey, CAR T cells must be able to detect chemokines at the tumor site, effectively roll and adhere to the blood vessel wall, initiate transendothelial migration, and invade the tumor stroma. Here, the immune cells must overcome various biochemical and physical barriers to then encounter pro-tumor cells that suppress CAR T cell activity. Some CAR T cells may eventually make contact with target cancer cells that express abundant immune checkpoints, further reducing anti-tumor function.
CAR T cells must traverse the endothelial junction and navigate through the tumor stroma that is inherently armed with abundant immunosuppressive factors to reach target cancer cells. The TME has long been identified as an active contributor to cancer progression
[6][5]. In this neoplastic microenvironment, different cell types interact with each other and the surrounding ECM to orchestrate a constantly evolving habitat that is favorable to resilient tumor growth
[7][8][9][19,20,21] and deleterious to immune function
[10][11][12][13][22,23,24,25] (
Figure 2). For instance, CAR T cells—upon successful infiltration of the TME—demonstrated a rapid loss of function
[14][26]. Infiltrating CAR T isolated from xenograft tumors and cultured in vitro for 24 h regained killing ability superior to those freshly isolated
[14][26]. This emphasizes the immunosuppressive influences of the TME that suppress CAR T anti-tumor activity. Furthermore, the stromal, immune, and cancer cells constantly remodel the TME in response to the surrounding biochemical and biophysical cues
[7][15][16][17][18][19,27,28,29,30] (
Figure 2). Unlike hematologic malignancies—which can be freely accessed by CAR T cells—solid tumors have a complex three-dimensional structure wherein malignant cells are more difficult to access
[19][20][21][22][31,32,33,34]. One hallmark of tumor progression is the elevated stiffening of the ECM due to increased matrix crosslinking, collagen deposition, and fiber alignment which regulate cell migration, proliferation, and apoptosis via mechanotransduction
[23][24][25][26][27][35,36,37,38,39]. The stiffening effect is often caused by collagen deposition and crosslinking, mediated by increased secretion of lysyl oxidase (LOX)
[18][28][30,40] and overproduction of other ECM components such as heparan sulfate proteoglycans (HSPGs)
[29][30][41,42]. Due to this physical constraint, primary tumor growth and cell migration within the TME are greatly dependent on matrix metalloproteinases (MMP) and heparinase enzymes to degrade and reorganize the crosslinked networks
[31][32][33][34][35][36][43,44,45,46,47,48]. However, T cells do not often secrete enzymes to degrade ECM, but rather choose the path of least resistance
[37][49] or follow contact guidance imposed by architectural features of the surrounding ECM
[38][39][50,51]. Therefore, densely packed and oriented stromal fibers have been reported to impose significant challenges to T cell infiltration
[40][52].
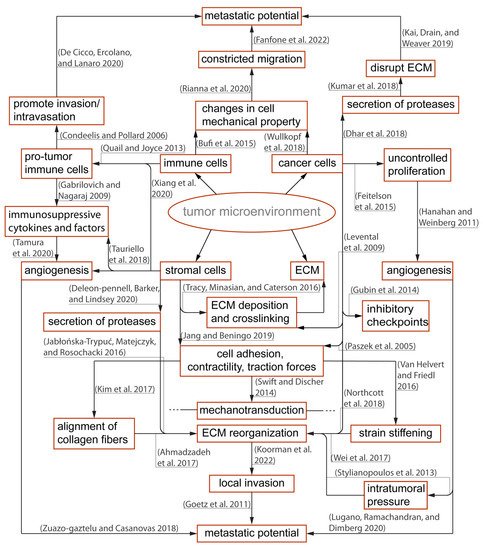
Figure 2. Overview of the immunosuppressive tumor microenvironment (TME) and important contributors to tumor progression: Within the TME, there is a dynamic relationship between peritumoral and intratumoral components that include immune cells, cancer cells, and the stromal elements often exhibiting context-dependent functionality. Different components of the TME carry out pro- or anti-tumor functions, and such polarizations are caused by reciprocal interactions with physical and biochemical cues from the surroundings. The polarized functions are not distinctive but rather heterogenous contributing to an immunosuppressive TME
[6][7][8][16][17][23][27][28][34][36][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][5,19,20,28,29,35,39,40,46,48,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75].
Tumor growth is critically dependent on angiogenesis
[15][64][65][27,76,77] which is promoted in an MMP–dependent manner
[31][43]. Given nutrient support from neovessel networks, the growing tumor continues to modify its microenvironment through elevation of growth-induced solid stress
[66][67][78,79], interstitial fluid pressure
[68][69][80,81], enzymatic secretion
[28][31][40,43], and cellular contractility onto the ECM
[56][59][68,71]. One example of ECM modification is the physical alignment of collagen fibers due to cell-induced strain stiffening
[16][57][70][28,69,82] from cancer cells and their stromal neighbors such as cancer-associated fibroblasts (CAFs)
[16][52][55][28,64,67]. This reorganization further stiffens the TME, suppresses immune activity and mobility
[71][72][83,84], and promotes local invasion of cancer cells along the aligned fibers
[43][61][71][73][74][75][76][55,73,83,85,86,87,88]. In addition, intratumoral pressure caused by growth-induced solid stress and interstitial fluid pressure obstruct delivery of therapeutic agents and trafficking of immune cells to the solid tumors
[77][89]. Elevated interstitial fluid pressure—reported in most solid cancers
[78][90]—is mainly caused by leaky blood vessels, impaired lymphatic transport, and hyaluronan swelling
[60][79][72,91]. The fluid pressure contributes to an increasing growth-induced solid stress and presents a non-trivial barrier to immune infiltration and uptake of therapeutic agents
[78][80][90,92]. An in vitro study demonstrated that an interstitial fluid pressure ≥ 1 KPa—simulated by hydrostatic pressure—is sufficient to physically hindered antigen-specific T cell infiltration into tumor site
[81][93]. Furthermore, growth-induced solid stress (≥2 kPa) —while impeding proliferation of cancer cells
[66][82][78,94] —can collapse blood vessels
[60][72] and contribute to vasculature abnormalities and subsequent increase in interstitial pressure
[59][71]. The reciprocal relationship further escalates total intratumoral pressure, exacerbates hypoxia, and increases pH and metabolic waste accumulation. TME determinants—such as hypoxia
[83][95] and TGFβ
[51][63]—stimulate the production of ECM
[11][53][84][85][23,65,96,97] and LOX
[86][98] to further reinforce the physical barriers that impede CAR T infiltration
[21][87][33,99]. The feedback loop continually contributes an immunosuppressive TME responsible for tumor pathogenic traits, its progression
[28][88][89][90][40,100,101,102], subtle microevolution
[91][92][93][94][95][103,104,105,106,107], and metastasis
[61][62][63][96][97][98][99][73,74,75,108,109,110,111]. To overcome these challenges, therapeutic agents for vascular normalization
[100][112] and degradation of ECM components—such as hyaluronidases
[101][113], collagens
[102][114], and LOX inhibition
[103][115]—have been developed to alleviate intratumoral pressure and improve perfusion transport of therapeutic agents and immune cell infiltration
[104][116].
2. Immunosuppressive Chemokines: The Invisible Barrier against CAR T Infiltration
The recruitment of CAR T cells to the TME, upon systemic or regional administration, depends largely on efficient chemotactic migration. Without biochemical hints, the probability that CAR T cells localize and migrate to a solid tumor would be low. Within a TME, the local accumulation of suppressive chemokines and cytokines—such as CXCL12
[105][106][117,118], TGFβ
[51][107][63,119], IL-10
[108][109][110][120,121,122], IL-4
[111][123], IL-35
[112][113][124,125] and other factors (e.g., reactive oxygen species (ROS), lactate
[114][126], prostaglandin E2 (PGE2), and adenosine
[115][127])—constitute a first major barrier of immune suppression. The ability to sense and migrate towards a tumor-specific site is a critical prerequisite to immune extravasation and infiltration; however, as a physiological protective mechanism, tumors produce insufficient chemoattractant ligands for T cells. Therefore, there have been significant efforts to increase CAR T chemotaxis and extravasation that endow the local delivery of favorable chemokines and cytokines (e.g., CXCL11
[116][128], RANTES, and IL-15
[117][129]); these efforts include the use of intratumorally delivered oncolytic virus to improve CAR T cell recruitment and anti-tumor activity with resulting better survival
[116][117][118][128,129,130]. Alternatively, there have been promising strategies that employ CAR T cells as cytokine carriers to promote infiltration and maintenance of a healthy TILs population. CAR T cells—engineered to secrete proinflammatory cytokines, IL-12
[4][119][17,131], IL-18
[120][132], IL-7
[121][133], and CCL19
[122][134] —have been reported to augment autocrine stimulation and potentiate paracrine signaling advantageous for survival, persistence, and recruitment of endogenous immune cells.
To enhance locomotion in the TME, CAR T cells are engineered to express receptors specific to tumor-derived chemokines. This approach has shown efficient chemotactic migration and demonstrated remarkable tumor regression in both in vitro and in vivo models. T cells transduced with a retroviral vector to express CXCR2, a receptor of chemokine CXCL1, were able to enhance direct migration toward tumor-derived chemokine
[123][135]. In another study, CCR2—a receptor of chemokine CCL2, co-expressed with a chimeric antigen receptor targeting tumor antigen GD2
[124][136] (expressed on neuroblastoma) and mesothelin
[125][137] (expressed on malignant pleural mesotheliomas)—demonstrated a significant increase (more than 10-fold) in homing and anti-tumor activity.
The researcheOur
s' group, Jin et al., has developed CD70-directed CXCR1 or CXCR2-modified CAR T cells that can co-opt IL-8 with robust antitumor activity and long-lasting immunologic memory against glioblastoma, ovarian, and pancreatic cancer xenograft models
[126][138]. This approach has received FDA IND approval soon to be evaluated in a first clinical trial in humans
[126][127][138,139]. Furthermore, CAR T cells have been designed to co-express dominant-negative receptors of factors (e.g., TGFβ
[128][140], PD-L1
[129][141]) to resist immune-inhibitory signals in the TME or co-express inverted cytokine receptor
[111][130][123,142] to reverse suppressive cytokine functions and stimulate anti-tumor activity. In a preclinical study for breast cancer, CAR T cells targeting MUC1 co-expressed with an inverted cytokine receptor comprised of an IL-4 exodomain linked to an IL-7 endodomain reversed inhibitory signals from IL-4 present in the TME and demonstrated durable T cells infiltration and memory formation
[131][143].
3. Pro-Tumor Stromal and Immune Cells Are Active Contributors to Immune Suppression
Once CAR T cells invade chemorepellent and physical barriers of the TME, they encounter stromal and immune cell populations that often exhibit context-dependent functionality. Cancer-associated fibroblasts (CAFs) are present in all solid tumors and represent a major component of the reactive tumor stroma integral to the development and maintenance of a complex TME
[132][144]. CAFs are responsible for abundant production of ECM, crosslinking enzymes, and cytokines that fortify the tumor and constitute a complex and heterogenous barrier against immune attacks
[133][145]. CAFs are major contributors to the production of immunosuppressive cytokines, such as TGFβ, which have also been shown to interfere with T cell migration and infiltration into tumors
[51][134][63,146]. CAFs have been reported to cross-present antigen and kill CD8+T cells in an antigen-dependent manner via PD-L2 and FASL expression
[135][147] and recruit other inhibitory immune cells to participate in suppressing anti-tumor function
[49][61]. Direct immunotherapeutic attenuation of CAF activity shows promising improvement of T cell trafficking and infiltration
[136][148]. Kakarla et al. developed CAR T cells targeting fibroblast activation protein (FAP), demonstrating a significant reduction in FAP-positive stromal cells and tumor growth in murine models. A combined application of FAP-CAR T and tumor-antigen CAR T cells resulted in anti-tumor activity superior to treatment with either CAR T alone
[137][149]. Despite generally being known for pro-tumor function, the role of CAFs in tumor progression is not totally understood. Depletion of CAFs accelerated pancreatic ductal adenocarcinoma progression and reduced survival in transgenic mice models
[138][150]. Although each cellular component of a TME exhibits a supportive role in tumor progression, their existing anti-tumor functions further complicate therapeutic strategies.
Tumor initiation and progression lead to inflammatory reactions that trigger recruitment and repair mechanisms by innate and adaptive immune response
[139][151]. In the complex TME, cancer, stromal, and immune cell populations continuously evolve. The pro-tumor immune subsets within the TME generally comprise tumor-associated macrophages (TAM)
[44][140][56,152], myeloid-derived suppressor cells (MDSCs), tumor-associated neutrophils (TAN), and regulatory T cells (Treg)
[42][50][141][54,62,153]. Macrophages and dendritic cells (DCs) are important myeloid cells of the innate immune system. TAM often adopt an M2- macrophage phenotype
[142][154] which is anti-inflammatory and pro-tumorigenic
[143][144][155,156]. TAMs secrete CCL2, which recruits and activates additional TAMs. M2 TAMs have revealed pro-tumor functions and immune suppressive effects via secretion of epidermal and angiogenic factors, IL10 and TGFβ
[145][157]. CAR T cells targeting M2-like TAMs in murine ovarian carcinoma, colon adenocarcinoma, and melanoma models reprogrammed the TME with enrichment of pro-inflammatory monocytes and activated CD8+T cells
[146][158].
MDSCs, a heterogeneous population of immature myeloid cells, are recruited into the tumor by various chemokines CCL1, CCL2, CCL5, or CXCL5
[141][147][153,159]. MDSCs can be divided into two major subtypes: granulocytic and monocytic. Granulocytic MDSCs secrete reactive oxygen species (ROS), whereas monocytic MDSCs produce increased levels of nitric oxide derivatives resulting in decreased T cell immune responses
[48][148][60,160]. A MUC1-directed CAR T cell approach concomitantly targeting tumor necrosis factor-related apoptosis-inducing ligand receptor 2 (TR2) expressed on MDSCs demonstrated enhanced antitumor tumor activity in breast cancers enriched with MDSCs and TME remodeling
[149][161]. In addition, there is a growing body of data on the anti-antitumor immunity of TANs. The role of TANs seems to vary based on the type of solid tumors
[150][162]. It has been described that TANs recruit MDSCs, TAMs, and Tregs to TME through secretion of IL-4, IL-10, arginine-1, and ROS which have inhibitory effects on cytotoxic T cells
[150][162].
Tregs are cells born in the thymus and express CD4+ FoxP3. Tregs within the TME can act antagonistically to effector T cells by secreting a number of cytokines (e.g., IL10, IL35, TGFβ), upregulating cytotoxic T lymphocyte antigen 4 (CTLA4), and inhibiting CD80/CD86 co-stimulatory pathways, by competing for binding of IL-2
[151][163]. An agonist antibody specific against glucocorticoid-induced TNFR-related receptor (GITR) decreased Treg mediated immunosuppression which correlated with augmented anti-glioblastoma immune response
[152][164]. Once in the TME, CAR T cells must overcome checkpoint inhibitory signals. Checkpoints (e.g., PD-L1, GITR) are physiological brakes that prevent autoimmunity
[146][147][148][158,159,160]. However, cancer cells abuse such mechanisms to avoid immune surveillance, and the complex TME further supports cancer cells by employing multifactorial and reciprocal pathways advantageous to immune suppression. For instance, IFNγ release during CAR T cell activation and killing of tumor cells induces upregulation of immune checkpoint proteins (e.g., PD-L1) which attenuate the anti-tumor function of cytotoxic T cells
[153][165]. On the other hand, cancer-associated cells can be reprogrammed to target cancer cells. Reinhard et al. revealed nanoparticles delivering RNA encoding for claudin 6—a developmental antigen—can transduce antigen presenting cells to express the immunological targets and enhance CAR T expansion and tumor regression in a mouse model
[154][166].
4. Physical Confinement and Mechanical Properties of Cell Nucleus Modulate T Cell Locomotion
T cell locomotion within the TME is an important aspect of immuno-oncology and has been extensively studied
[38][39][155][156][50,51,167,168]. T cells are capable of dynamically adapting to various modes of migration depending on ECM composition within the TME
[38][156][157][50,168,169]. However, the mechanism behind this adaptation has not been clearly explained. A better understanding of T cell migration will help enhance CAR T trafficking to solid tumors. The modes of cell migration are often regulated by the density of binding sites on substrate
[158][170], proteolytic activity
[159][171], actomyosin contractility
[38][160][50,172], microtubule stability
[39][51], and geometrical and mechanical properties of the confined surroundings
[39][41][155][157][161][51,53,167,169,173]. T cells are fast migrators, for they need rapid scanning abilities to carry out efficient immunological response. Two-photon microscopy imaging of intact mouse lymph nodes revealed that T cells can migrate at an average speed of 10 µm min
−1 and peak at 25 µm min
−1 [162][174]. Unlike cancer and stromal cells that actively secrete protease enzymes to degrade the ECM
[47][59] and migrate via focal adhesion mediated attachment, T cells do not need mature focal adhesions and prefer to explore the TME via paths of least resistance
[156][163][164][168,175,176]. In other words, leukocytes probe their surroundings and selectively squeeze through accessible pores without proteolytically breaking down ECM to create paths for migration. Although capable of integrin-dependent migration, T cells only form short-lived adhesion complexes to facilitate rapid detachment and engagement onto the next location
[165][177]. The most common mode of T cell migration is amoeboid which employs coordinated membrane extrusion, cell contractility, and contact-induced traction in confined spaces to move forward
[156][165][168,177]. Amoeboid migration is critically dependent on cortical contractility regulated via the Rho/ROCK pathway and myosin II activity
[38][50]. It has been reported that inducing actomyosin contractility by increasing myosin II or activation of the Rho/ROCK pathway is sufficient to transform cell mode of migration into amoeboid
[160][166][167][172,178,179]. Besides cell contractility, proper geometrical confinement is an essential determinant of amoeboid migration
[157][169]. T cells protrude oscillatory leading-edge membrane blebs into narrow pores, probe for potential paths, and generate contraction-mediated retrograde actin flow beneath the cell membranes to transmit equal and opposite frictional forces onto the substrate to drive the cell forward
[38][156][50,168].
The compliance of the cells can better facilitate constricted migration
[43][55]. The deformability of a cell depends significantly on the mechanical property of its nucleus
[168][169][180,181]. During transendothelial migration through tight spaces, CAR T may be subjected to nuclear rupture which could severely impact cell function
[170][182]. Major determinants of nuclear stiffness are lamin A/C content and chromatin decondensation which are mechanically regulated by force transmission between cell–cell and cell–matrix interaction via cytoskeleton and LINC complex
[58][70]. Lamin A is known to be responsible for the viscous portion of the nucleus which leads to plastic deformation post-migration, whereas lamin B is responsible for the elastic portion promoting shape recovery after deformation
[171][172][183,184]. Although high lamin A expression impedes migration through small pores (<3 µm in dia.), it promotes cell survival against stress-induced apoptosis by upregulation of DNA damage repair protein, HSP90
[173][174][185,186].
Furthermore, cells are well-known mechanosensors
[24][58][175][176][177][178][179][36,70,187,188,189,190,191] and can alter their mechanical properties and mode of migration according to the stiffness of local surroundings
[39][43][45][46][51,55,57,58]. On a soft substrate, lamin A/C phosphorylation is increased; whereas on stiff substrate, myosin II-generated tension promotes dephosphorylation and stabilization of lamin A/C levels which in turn enhance nuclear stiffness
[180][192]. Moreover, mechanotransduction is a key regulator in T cell activation
[181][182][193,194] and cytotoxic activity
[179][191]. In the context of T cell migration, the mechanical stiffness of the substrate is important for durotaxis and contact guidance-directed locomotion in 3D
[39][51]. T cells apply traction forces (100 pN) via TCR to physically probe the rigidity of their surroundings
[177][189] and migrate along with the aligned ECM fibers
[183][195]. Although matrix optimal confinement and alignment could induce rapid T cell amoeboid migration, there is a limit to which cells are physically trapped and suppressed. For instance, high-density collagen deposition and crosslinking have been shown to suppress proliferation and anti-tumor activity of infiltrating CD8+ T cells in triple-negative breast cancer tissue explants
[72][84]. Another study shows that a densely packed and oriented stromal fibers at the perivascular niche and around the tumor impose a significant challenge to T cell infiltration
[40][52]. Recently, Caruana et al. developed CAR T cells expressing heparinase to degrade HSPGs and promote tumor infiltration. In xenografted mouse models, the strategy improved T cell capability to degrade ECM, infiltrate, and carry out antitumor activity
[35][47].