Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Carla Fernandes and Version 2 by Vivi Li.
Oceans are a rich source of structurally unique bioactive compounds from the perspective of potential therapeutic agents. Marine peptides are a particularly interesting group of secondary metabolites because of their chemistry and wide range of biological activities. Among them, cyclic peptides exhibit a broad spectrum of antimicrobial activities, including against bacteria, protozoa, fungi, and viruses. Moreover, there are several examples of marine cyclic peptides revealing interesting antimicrobial activities against numerous drug-resistant bacteria and fungi, making these compounds a very promising resource in the search for novel antimicrobial agents to revert multidrug-resistance.
- marine peptides
- cyclic peptides
- cyclic depsipeptides
- antimicrobial resistance
- peptide synthesis
1. Introduction
Despite great advances in the pharmaceutical and medicine fields, contagious diseases induced by bacteria, fungi, viruses, and protozoa are still a significant threat to public health, as evidenced by the SARS-Cov-2 pandemic [1]. Due to the emergence of new pathogenic agents, extensive resistance, and the lack of new drugs, contagious diseases affect both developed and developing countries [2][3][2,3].
The golden age of antibacterial agents began in the 1940–1960s, and many antibiotics dating from that period are still used in therapy today. Due to the high rate of antibiotics discovery, during this period, it was believed that infectious diseases would soon be controlled in the population [4]. A line of research on antimicrobial discovery and development was identified from many natural small-molecule products that were clinically proved to have antibacterial activity. Among these small molecules, penicillins, cephalosporins, macrolides, glycopeptides, tetracyclines and aminoglycosides stand out. On the other hand, another line of research was found from the structures of the chemical dye industry, leading to the discovery of aromatic sulfa compounds with antibiotic activity [5]. In 1960, the fluoroquinolones emerged, which are the second example of synthetic antibiotics used in therapy. Later, in the 2000s, the first generation of oxazolidinone linezolid, a synthetic derivative structurally different from the previous ones, was approved in the USA [6]. In addition, new generations of cephalosporins, macrolides, fluoroquinolones, and tetracyclines appeared with significant therapeutic use. It is important to highlight that the development of new synthetic methodologies has allowed the synthesis of pentacyclines, derived from tetracyclines, which may be considered as a fourth generation of this class of antibiotics [7].
Regarding the treatment of systemic mycoses, such as candidiasis, aspergillosis, and cryptococcosis, antifungals can be organized into four classes—polyenes, azoles, flucytosine, and echinocandins—in which they are distinguished by the mode of formulation, bioavailability, pharmacological interactions, adverse effects, and mechanism of action [8][9][8,9]. Although commensals in humans, Candida species are a cause of various infections in susceptible patients, including elderly, hospitalized, and immunosuppressed patients. Invasive Candida infection is one of the most common fungal infections globally [10]. Less common, but responsible for greater treatment concerns, are systemic infections caused by fungi of the genera Aspergillus, Fusarium and Scedosporium, considering their susceptibility profile to the available antifungals [11][12][11,12].
Over the course of human civilization, viral infections have caused millions of human casualties worldwide, driving the development of antiviral drugs in a pressing need [13]. As of April 2016, antiviral drugs have been approved to treat nine human infectious diseases: hepatitis B, hepatitis C, and infections caused by human immunodeficiency virus (HIV), human cytomegalovirus, herpes simplex virus (HSV), human papillomavirus, respiratory syncytial virus, varicella-zoster virus, and influenza virus [14][15][14,15]. Nevertheless, there is still no antiviral drug or vaccine for over 200 human viruses afflicting populations worldwide [14]. In addition, the current pneumonia outbreak caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was declared a pandemic by the World Health Organization on 11 March 2020 [16].
Parasitic diseases are a critical health concern with a profound impact on the global human population [17]. It was found that protozoans, such as Trypanosoma cruzi, Leishmania mexicana, Plasmodium falciparum, Giardia intestinalis, and Trichomonas vaginalis, are the major disease-causing parasites. The spread of infectious diseases is especially prevalent in underdeveloped countries characterized by tropical or temperate climate, as well as poor sanitary and hygienic conditions [18][19][20][18,19,20]. Parasite infections are the cause of 500 million deaths worldwide [21][22][21,22], and although there are drugs to fight parasite infections, these have drawbacks such as toxicity and the emergence of resistance [23][24][23,24].
Antimicrobial resistance (AMR) is another significant threat to public health systems all over the world [25]. Infection caused by microorganisms resistant to antimicrobial drugs leads to serious illnesses and elevated costs associated with more expensive antibiotics (when infections become resistant to first-line antimicrobials, treatment has to be switched to second- or third-line drugs, which are nearly always more expensive), specialized equipment, longer hospital stay, and isolation procedures for the patients. Societal costs include loss of productivity and death [25][26][25,26]. Every year, more than 2 million North Americans acquire infectious diseases associated with antibiotic-resistant microorganisms, resulting in 23,000 deaths [27]. In Europe, nearly 700 thousand cases of antibiotic-resistant infections develop directly into over 33,000 deaths yearly [28]. Despite a 36% increase in human use of antibiotics from 2000 to 2010 [29], approximately 20% of deaths worldwide are related to infectious diseases today [30]. Statistical models predicted that there were an estimated 4.95 million deaths associated with AMR in 2019, including 1.27 million deaths associated with bacterial AMR [3]. The six leading pathogens associated with AMR in 2019 (Escherichia coli, followed by Staphylococcus aureus, Klebsiella pneumoniae, Streptococcus pneumoniae, Acinetobacter baumannii, and Pseudomonas aeruginosa) were responsible for 3.57 million deaths [3].
AMR can occur through several mechanisms, whether intrinsic or acquired. Intrinsic resistance occurs naturally, as part of a microbial evolution process. Acquired resistance, on the other hand, is a consequence of the indiscriminate use of antimicrobials, and genetic mutations may occur, originating resistance genes that can be transferred between microbial species. AMR mechanisms fall into four main categories [31], as shown in Figure 1.
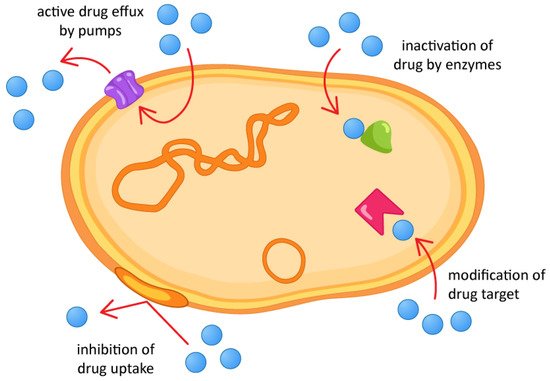
Figure 1.
Antimicrobial resistance mechanisms.
Ocean water covers about 70% of the Earth’s surface and contains several potential bioactive compounds to be discovered. Marine organisms have been considered to be a promising source of numerous nutraceutical and pharmaceutical compounds [32][33][32,33]. Over the last few decades, new marine-derived compounds have been considered not only as lead compounds for drug discovery, but also as bioactive agents in pharmaceutical research, possessing antifungal, antibacterial, cytotoxic and anti-inflammatory properties, among others [34][35][36][37][34,35,36,37].
Specific chemical and physical properties, such as water salt concentrations, pressure, temperature (including extreme), light penetration, ocean currents, oxygen concentrations, and radiation exposure characterize different underwater habitats (environment) of marine organisms [38][39][38,39]. Due to this extreme environment, marine organisms are forced to produce a chemical diversity of bioactive compounds that are considered essential for the discovery and development of new agents for the treatment and prevention of various fungal, bacterial, viral, and protozoal infections [35][40][41][42][43][44][45][35,40,41,42,43,44,45].
In particular, several peptides have been isolated from marine sources and demonstrated to be promising drug candidates based on their significance of the bioregulatory role and unique molecular mechanisms of action [46][47][46,47]. ReseaOurchers' group also described the isolation and stereochemical analysis of marine peptides [48][49][48,49]. Compared to small-molecule drugs, peptides are highly selective and efficacious and, at the same time, relatively safe and well tolerated [50]. The high degree of selectivity in their interactions is the result of millions of years of evolutionary selection for complementary shapes and sizes from among a large array of structural and functional diversity [47].
Despite the applicability of peptides and proteins in medicine, it has been limited by their high manufacturing costs, the low bioactivity of peptides when administered orally, and their low membrane permeability, low systemic stability, and high clearance rates [51]. The low stability under physiological conditions is one of the main obstacles to the therapeutic use of linear peptides, as they easily lose their biological activity because they are rapidly cleaved by enzymes in vivo [52][53][52,53]. To overcome this obstacle, diverse peptide modifications have been proposed [54][55][56][57][54,55,56,57]. Linear peptide cyclization has recently been considered one of the most promising approaches, due to several advantages in surpassing both pharmacokinetic and pharmacodynamic limitations. A cyclic structure reduces the conformational freedom for each constituent within the cycle and forces the molecule into a more rigid secondary structure [58]. The increase in rigidity is an advantage that is translated into a decrease in the entropic term of Gibbs energy, improving binding affinities higher than some natural ligands to a biotarget [59][60][61][62][59,60,61,62]. Common motifs in the formation of proteins and polypeptides, β-turn, are other advantages of cyclization, as it is believed that this improves binding affinity [63][64][65][63,64,65]. Cyclization also allows the elimination of charged terminals at the ends of the structure in cyclic peptides, which may increase membrane permeability [66], although the peptide cross of the membrane does not improve just because the peptide is cyclized, but due to its structural features [67]. Another advantage of cyclization is becoming less prone to hydrolysis, as it decreases the exposure of the amino and carboxyl termini to exopeptidases [68], decreasing off-target side effects [69], thus leading to substantially enhanced metabolic stability and specificity [70][71][72][73][70,71,72,73]. Furthermore, in terms of chemical synthesis, cyclic peptides are significantly smaller when compared to proteins, and therefore, lower manufacturing costs are needed [74]. Actually, cyclic peptides are polypeptide chains that contain a circular sequence of bonds, which can be formed through a bond between the amino and carboxyl termini of the peptide with an amide bond, or other chemically stable bonds such as lactone, ether, thioether, and disulfide, among others. The formation of the amide bond between the amino and carboxylic terminals results in the formation of a head-to-tail (or N-to-C) cyclic peptide. Many cyclic peptides with this kind of formation (N-to-C) exist in nature [75][76][77][78][79][75,76,77,78,79].
Depending on lipophilicity, the type of bonds between amino acids, and the number of amino acids, cyclic peptides can have different classifications, as either cyclic lipopeptides, cyclic glycopeptides, or cyclic depsipeptides. Cyclic lipopeptides are cyclic peptides acylated by a lipid, usually a fatty acid (FA) side chain. These compounds are produced only in bacteria and fungi of various habitats during cultivation on carbon and nitrogen sources [80]. Cyclic glycopeptides contain carbohydrate moieties covalently attached to the side chains of amino acid residues [81]. Cyclic depsipeptides are cyclic peptides in which amide groups are replaced by corresponding lactone bonds due to the presence of a hydroxy-carboxylic acid in the peptide structure by cyclization to the hydroxyl of serine or threonine side chains [82]. Modification of the amide bond to an ester increases the lipophilicity that may subsequently improve cell permeability. Depsipeptides have also been used to demonstrate the importance of the hydrogen bonds that are formed by amide bonds in natural peptides [79]. These peptides sometimes display additional chemical modifications, including unusual amino acid residues in their structures. Cyclic depsipeptides contain at least one ring formed only through peptide or ester links—derived from hydroxy carboxylic acids [83]. Cyclic peptides and cyclic depsipeptides can be named as cyclic tri-, tetra-, penta-, hexa-, hepta-, octa-, nona-, deca-, unde-, dodeca, and tri-decapeptides, depending on the number of amino acids present [84].
Depsipeptides are also called peptolides or nonribosomal peptides (NRP), being biosynthesized by non-ribosomal peptide synthetases (NRPS) in combination with either polyketide synthase (PKS), or FA synthase enzyme systems, and are often found in marine organisms such as bacteria, tunicates, mollusks, and sponges, among others [76][77][78][76,77,78]. NRPS are multifunctional proteins that synthesize peptide natural products independent of the mRNA ribosomal machinery and employ a modular architecture wherein each module is responsible for the incorporation of one amino acid into the final peptide product [85][86][85,86]. NRP can be linear, cyclic, or branched peptides, and usually contain fewer than 20 amino acid residues and are often modified by chemical processes such as acylation, glycosylation, and others. Each module functions as a building block responsible for the incorporation and modification of an amino acid, especially D-amino acids, so that the order and quantity of modules in an NRPS determine the amino acid sequence of the synthesized peptide [87].
The section of the NRPS enzyme that specifically incorporates an amino acid into the peptide chain is defined as a module, and the modules, in turn, can be divided into domains, which catalyze the individual steps of non-ribosomal peptide synthesis. Each standard elongation module consists of three domains: a condensation domain (C), an adenylation domain (A), and a peptidyl transporter protein (PCP), organized as C-A-PCP [87] (Figure 2). The PCP domain is also frequently referred to as the T domain, and the holoform of this domain is a substrate for thioester (“thiolation”) formation [88][89][90][91][92][88,89,90,91,92]. In addition to these main domains, there are others involved in modifying an NRP: the E domain (molecule epimerization); the Cy domain (cyclization of the forming molecule); the MT domain (methylation reactions); the R domain (reduction reactions), and the Ox domain (oxidation reactions) [93].
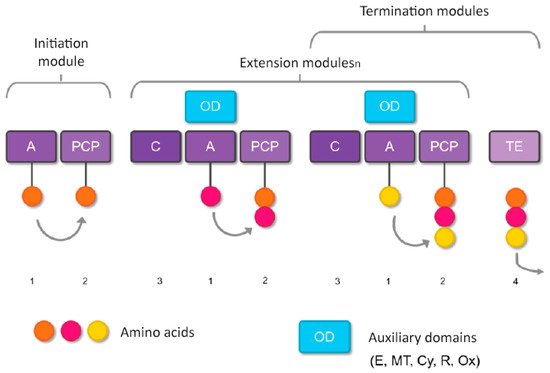
Figure 2. Basic steps of nonribosomal peptide synthesis. (1) Domain A selects the amino acid to be incorporated and transfers it to the PCP domain (2), where a thioester bond is formed. (3) Domain C forms the peptide bond between the amino acid present in the PCP domain of the same module and the intermediate peptidyl linked to the PCP domain of the previous module (that is, it catalyzes the link between amino acids of adjacent modules). (4) If no additional domains are present that modify the molecule during formation, the TE domain releases the formed peptide. However, if additional domains are present (such as E, MT, Cy, or Ox), the molecule is modified before being released by the TE domain.
The terminating domain, the thioesterase (TE) domain, normally releases the peptide by hydrolysis or cyclization [94], while reductase (R) domains catalyze release by converting the thioester link to an aldehyde [95], or alcohol [96], specialized C domains [97], catalyze cyclization [98], and amide bond formation through small molecule acceptance [97][99][100][101][97,99,100,101].
2. Marine Cyclic Peptides with Antimicrobial Activities
2.1. Sponge-Produced Cyclic Peptides
Sponges are diversified organisms distributed extensively on shores and deep in the ocean [102][112]. In terms of chemical diversity, an exceptionally prolific group of sponges include the lithistid sponges, the prominence of which may be due to the biosynthetic capacity of the microorganisms that host them [103][104][105][113,114,115]. The metabolites of lithistid sponges, which include the genera Theonella, Discodermia, Aciculites, Microscleroderma, and Callipelta, are among the most diverse found in any order of sponges and have often been attributed to symbiotic microorganisms (such as proteobacteria “Candidatus Entotheonella palauensis”) [104][114], that contains four distinct cell populations: sponge cells, unicellular heterotrophic bacteria, unicellular cyanobacteria, and filamentous heterotrophic bacteria. In particular, many cyclic peptides and peptide lactones have been reported to be able to be obtained from Theonella sponges, and their structural features, including unusual amino acids or D-amino acids, suggest that they perhaps originated from symbiotic microorganisms [106][107][116,117]. In this section, 63 cyclic peptides isolated from sponges (1–63) are described (Table 1 and Figure 3). Among these, 14 cyclic peptides have been found to have antibacterial activities, as well as 19 with antifungal, one with parasitic, and 22 with antiviral activities. The most relevant antimicrobial cyclic peptides are highlighted due to their unusual structural characteristics or advanced investigations, potency, and in vivo experiments.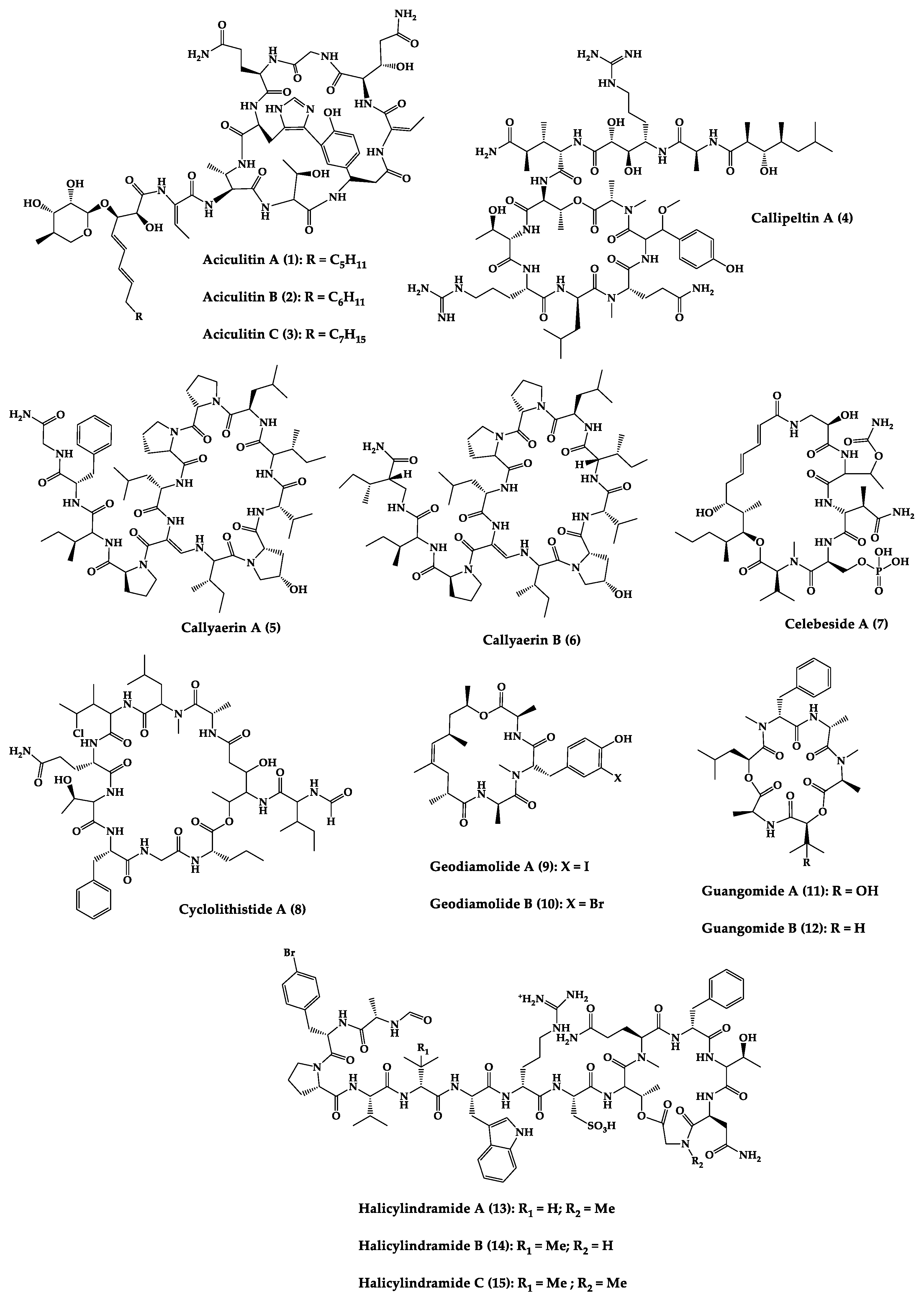
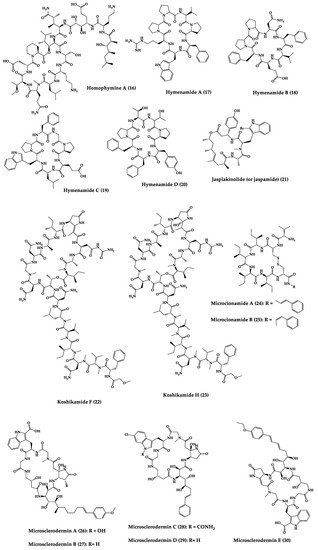
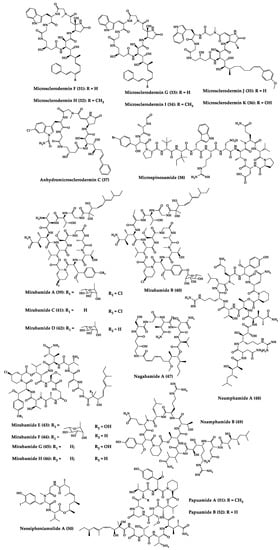
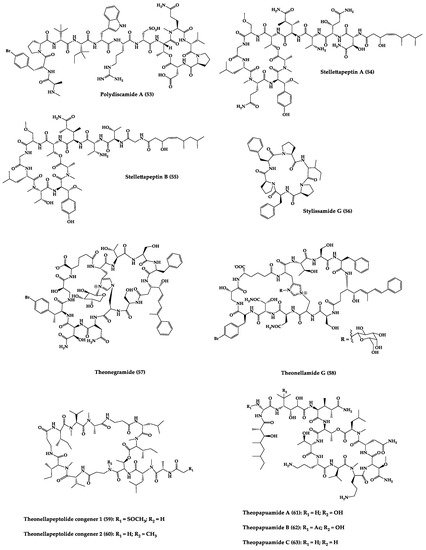
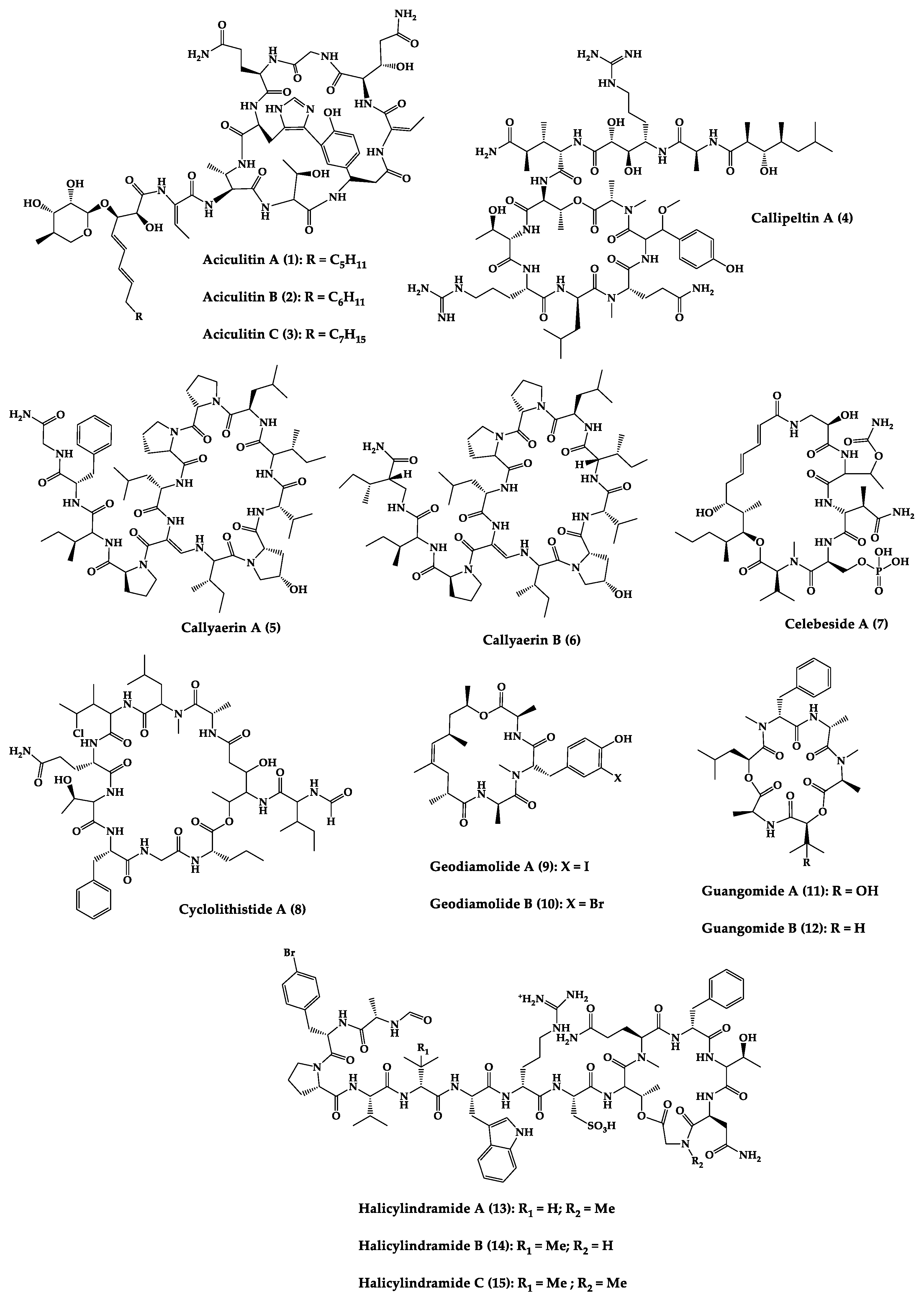
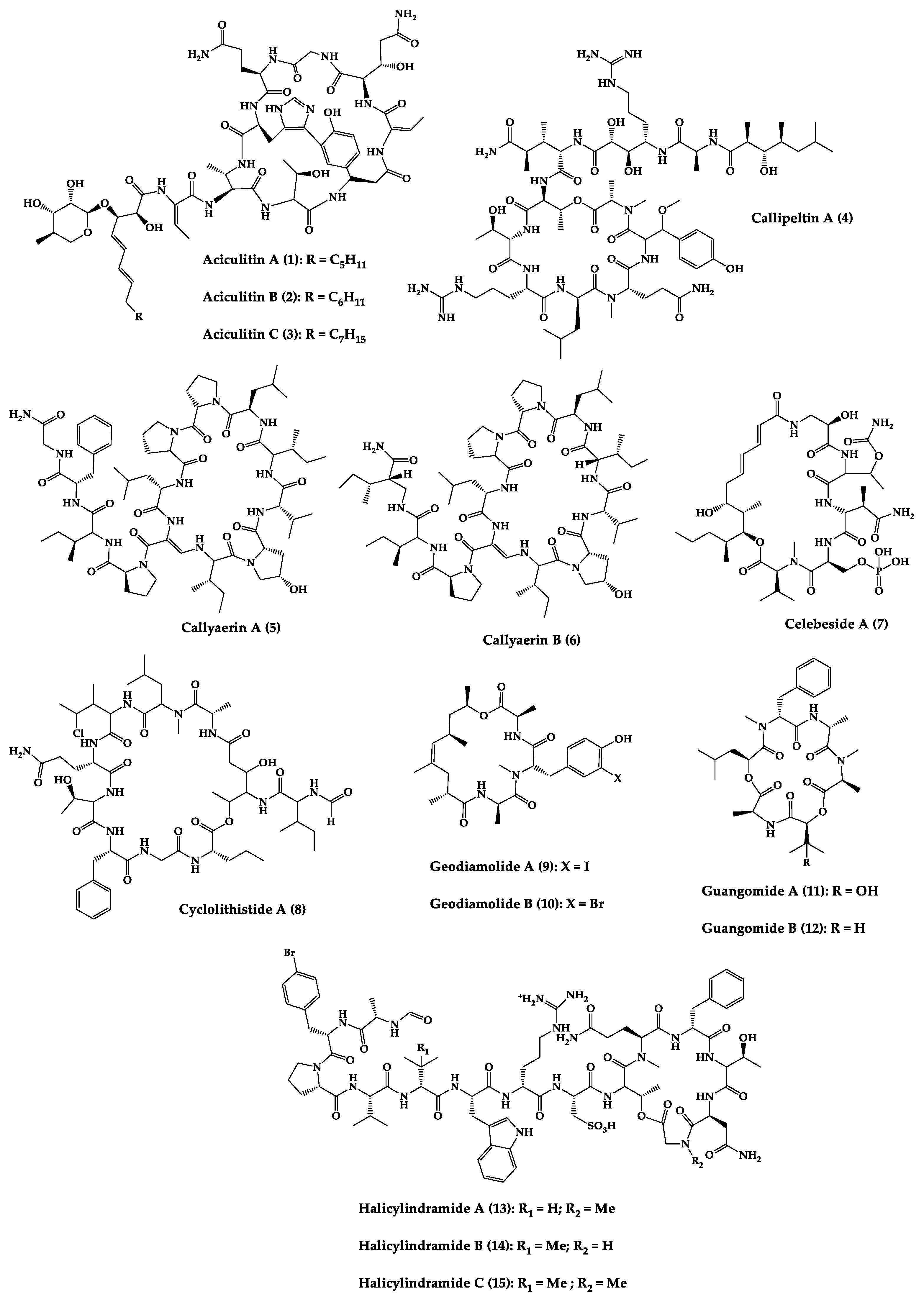
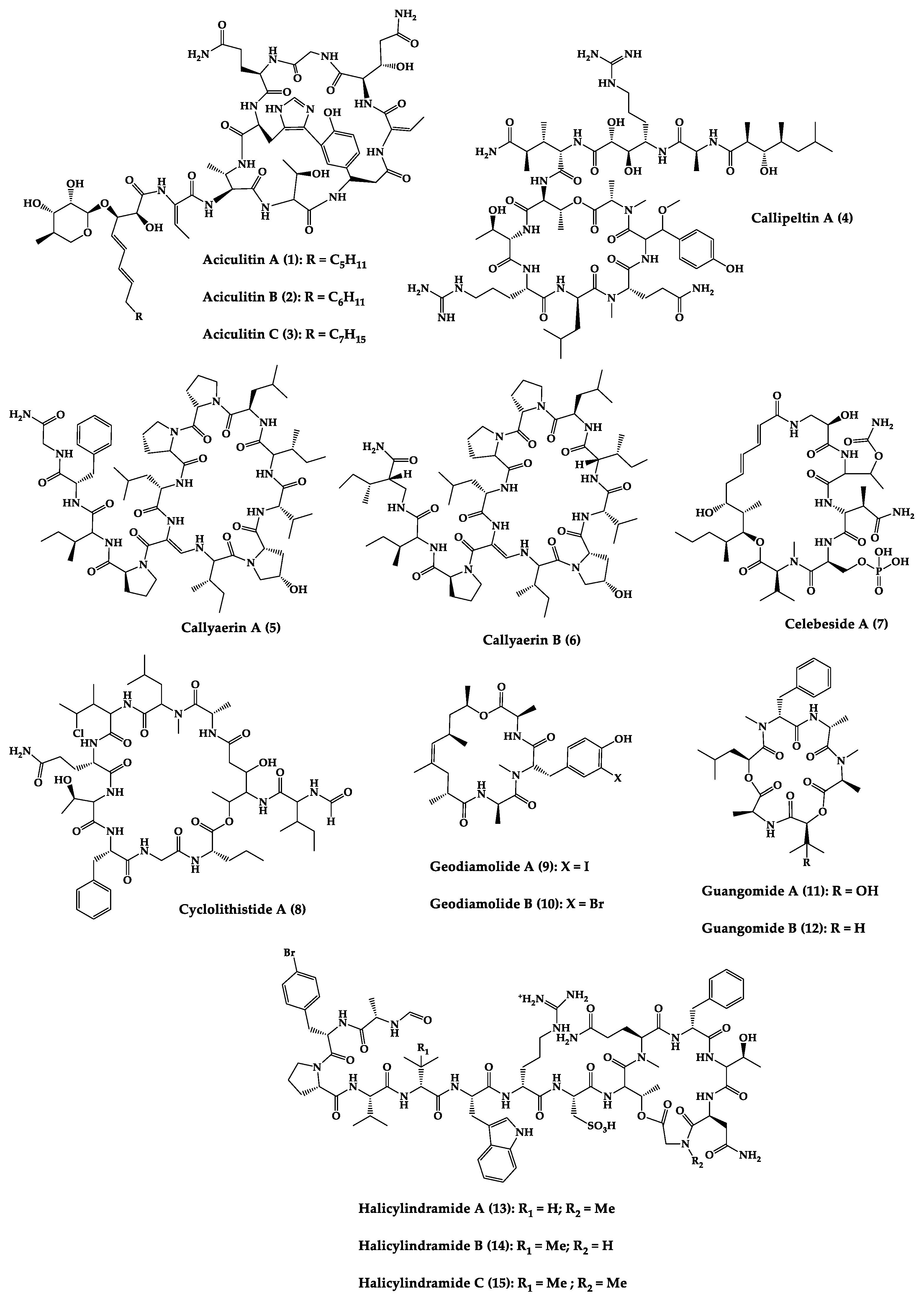
Figure 3.
Chemical structures of cyclic peptides from sponges (
1
–
63
).
Table 1.
Antimicrobial cyclic peptides from marine sponges.
| Compound | Structure | Source | Antimicrobial Activity | Synthesis | References |
|---|---|---|---|---|---|
| Aciculitins A-C (1–3) | Bicyclic octa-peptides | Aciculites orientalis | C. albicans (2.5 µg/disk, standard disk assay) | Semi-synthesis | [124][125][134,135] |
| Callipeltin A (4) | Cyclic deca-depsipeptide | Callipelta sp. | HIV-1 infection inhibition (CD50 = 0.29 µg/mL, ED50 = 0.01 µg/mL), C. albicans (100 µg/disk) | Total synthesis of analogues | [126][127][128][129][130][136,137,138,139,140] |
| Callyaerins A (5) and B (6) | Cyclic undeca-peptides | Callyspongia aerizusa | IC90: M. tuberculosis (2 μM and 5 μM, respectively), isoniazide (0.625 μM) | Total synthesis | [131][132][141,142] |
| Celebeside A (7) | Cyclic penta-depsipeptide | Siliquaria-spongia mirabilis | IC50: Neutralized HIV-1 (1.9 µg/mL) | - | [123][133] |
| Cyclolithistide A (8) | Cyclic deca-despipeptide | Bacteria symbiosis Theonella swinhoei | C. albicans (20 µg/disk) | - | [133][143] |
| Geodiamolides A (9) and B (10) | Cyclic depsipeptides | Geodia sp. | MIC: C. albicans (31.3 µg/mL) | Total synthesis | [134][135][136][144,145,146] |
| Guangomides A (11) and B (12) | Cyclic tetra-depsipeptides | Unidentifiable sponge derived fungus | MIC: S. epidermidis (100 µg/mL), E. durans (100 µg/mL) |
- | [137][147] |
| Halicylindramides A-C (13–15) |
Cyclic tetra-decapeptides | Halichondria cylindruta | M. ramanniana (7.5 µg/disk) | Total synthesis and analogues | [108][138][139][118,148,149] |
| Homophymine A (16) | Cyclic undeca-depsipeptide | Homophymia sp. | IC50: HIV-1 infection cytoprotective (75 nM) | Semi-synthesis | [109][140][141][119,150,151] |
| Hymenamides A (17), B (18), C (19), and E (20) | Cyclic hepta-peptides | Hymeniacidon sp. | MIC: C. albicans (33–66 µg/mL), C. neoformans (33–133 µg/mL) | Total synthesis and analogues | [142][143][144][152,153,154] |
| Jasplakinolide (or jaspamide) (21) | Cyclic depsipeptide | Jaspis sp. | H. virescens (LC50 = 4 ppm), N. brasiliensis (LD50 < 1 µg/mL), C. albicans (MIC > 25 µg/mL), in vivo murine vaginal C. albicans infection (2% jasplakinolide was equivalent in efficacy to administration of miconazole nitrate at 2%) |
Total synthesis and analogues | [110][111][112][145][146][147][120,121,122,155,156,157] |
| Koshikamides F (22) and H (23) | Cyclic heptadeca-peptides | Theonella swinhoei and T. cupola | IC50: HIV-1 neutralization (2.3–5.5 µM) | - | [45][113][45,123] |
| Microcionamides A (24) and B (25) | Cyclic hexapeptides | Clathria abietina | MIC: M. tuberculosis (5.7 µM) |
- | [148][158] |
| Microsclero-dermins A–K (26–36) and anhydromicros-clerodermin C (37) |
Cyclic hexapeptides | Cyanobacteria simbiosis Microsclero-derma herdmani sp. and Theonella sp. | C. albicans (2.5–100 µg/disk, standard disk assay) |
Total synthesis and analogues | [149][150][151][152][153][159,160,161,162,163] |
| Microspinosamide (38) | Cyclic trideca-depsipeptide | Sidonops microspinosa | EC50: HIV-1 infection inhibition (0.2 µg/mL) |
Semi-synthesis | [114][154][124,164] |
| Mirabamides A–H (39–46) |
Cyclic glyco-depsipeptides | Siliquarias-pongia mirabilis and Stelletta clavosa | IC50: neutralized and fusion HIV-1 (40 nM–3.9 µM), B. subtilis, C. albicans (1–5 µg/disk) |
Semi-synthesis | [115][116][155][125,126,165] |
| Nagahamide A (47) | Cyclic hexa-depsipeptide | Theonella swinhoei | E. coli or S. aureus (50 µg/disk, inhibition zone 7 mm) |
Semi-synthesis | [156][166[157],167] |
| Neamphamide A (48) | Cyclic undeca-depsipeptide | Neamphius huxleyi | EC50: HIV-1 infection cytoprotective (28 nM) | - | [118][128] |
| Neamphamide B (49) | Cyclic undeca-depsipeptide | Neamphius sp. | MIC: M. smegmatis (1.56 µg/mL), M. bovis (6.2–12.5 µg/mL) |
- | [119][129] |
| Neosiphoniamolide A (50) | Cyclic tetra-depsipeptide | Neosiphonia suprtes | P. oryzae (IC90 = 5 ppm) H. gramineum (MIC ≤ 2 µg/mL) | - | [158][168] |
| Papuamides A (51) and B (52) | Cyclic depsipeptides | Bacteria symbiosis Theonella mirabilis and Theonella swinhoei | EC50: HIV-1 infection inhibition (1–74 ng/mL) |
Total synthesis and analogues | [120][155][159][160][161][162][130,165,169,170,171,172] |
| Polydiscamide A (53) | Cyclic tridecapeptide | Discodermia sp. | MIC: B. subtilis (3.1 µg/mL) | Total synthesis and analogues | [163][164][173,174] |
| Stellettapeptins A (54) and B (55) | Cyclic undecadepsi-peptides | Microorganisms symbiosis Stelletta sp. | EC50: infection of human T-lymphoblastoid cells by HIV-1RF (23 and 27 nM, respectively) | - | [165][175] |
| Stylissamide G (56) | Cyclic heptapeptide | Stylissa caribica | MIC: M. audouinii, T. mentagrophytes, C. albicans (6 μg/mL) | Total Synthesis | [166][176] |
| Theonegramide (57) | Bicyclic glycododecapeptide | Bacteria symbiosis Theonella swinhoei | C. albicans (10 µg/disk) |
- | [167][177] |
| Theonellamide G (58) | Bicyclic glyco-depsipeptide | Bacteria symbiosis Theonella swinhoei | IC50: Wild and amphotericin B-resistant strains of C. albicans (2.0–4.49 μM), amphotericin-B (1.48 μM) | Semi-synthesis | [121][168][131,178] |
| Theonellapeptolide congeners 1 (59) and 2 (60) | Cyclic trideca-depsipeptides | Theonella sp. | MIC: S. aureus (8.0–16 µg/mL), M. luteus (8.0 µg/mL), B. subtilis (8.0–16 µg/mL), M. smegmatis (16–66 µg/mL), T. mentagrophytes (4.0–8.0 µg/mL), A. niger (8.0–66 µg/mL) | Total synthesis and analogues | [169][170][179,180] |
| Theopapuamide A-C (61–63) | Cyclic undeca-depsipeptides | Bacteria symbiosis Theonella swinhoei and Siliquarias-pongia mirabilis | Wild type and amphotericin B-resistant strains of C. albicans (1–5 µg/disk); in vitro HIV-1 infectivity assay IC50 = 0.8 μg/mL |
- | [123][133] |
CD50 (median convulsant); EC50 (effective concentration in 50% of population); ED50 (effective dose in 50% of population); HIV (human immunodeficiency virus); IC50 (half maximal inhibitory concentration); IC90 (maximum inhibitory concentration in 90% population); LD50 (lethal dose in 50% population); MIC (minimum inhibitory concentration). Aspergillus niger (A. niger); Bacillus subtilis (B. subtilis); Candida albicans (C. albicans); Cryptococcus neoformans (C. neoformans); E. coli (Escherichia coli); Enterococcus durans (E. durans); Heliothis virescens (H. virescens); Helminthosporium gramineum (H. gramineum); Micrococcus luteus (M. luteus); Microsporum audouinii (M. audouinii); Mortierella ramanniana (M. ramanniana); Mycobacterium species (M. bovis, M. smegmatis, M. tuberculosis); Nippo-Strongylus brasiliensis (N. brasiliensis); Piricularia oryzae (P. oryzae); Staphylococcus species (S. aureus, S. epidermidis); Trichophyton mentagrophytes (T. mentagrophytes).
2.2. Bacteria-Produced Cyclic Peptides
A great variety of bacteria can be found in different marine habitats. Recent studies have shown that the main phyla of marine bacteria have a wide range of inhibitory activity against different types of microorganisms. Many antimicrobial substances active in a wide range of target organisms are produced by marine bacteria [171][181]. In this section, 53 cyclic peptides isolated from bacteria (64–116) have been reported (Figure 4). Among these, 38 cyclic peptides have been described with antibacterial activities, as well as 14 with antifungal, five with antiparasitic, and one with antiviral activities. The most relevant antimicrobial cyclic peptides are highlighted here due to their unusual structural characteristics or advanced investigations, potency, and in vivo experiments.
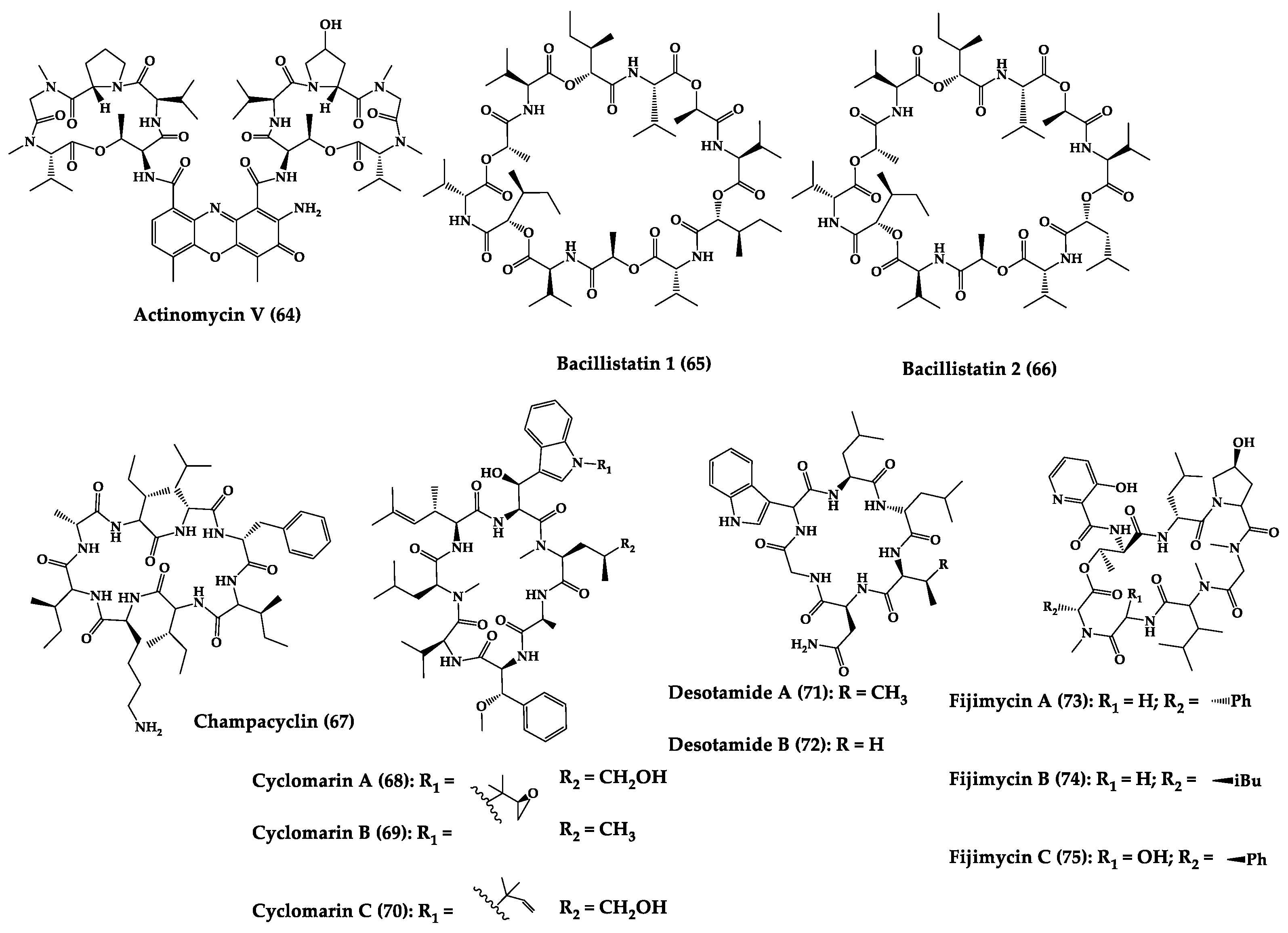

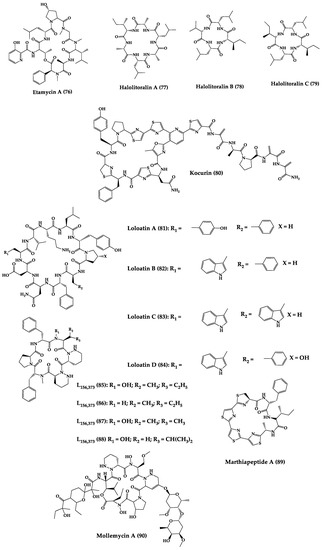
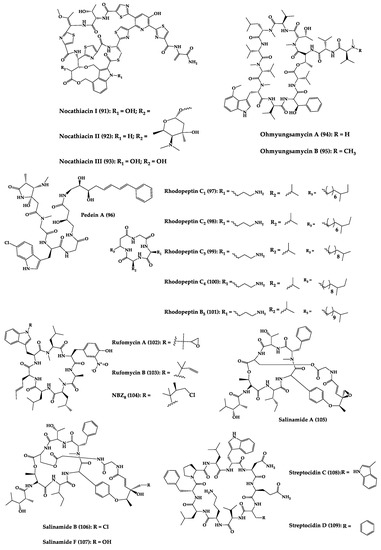
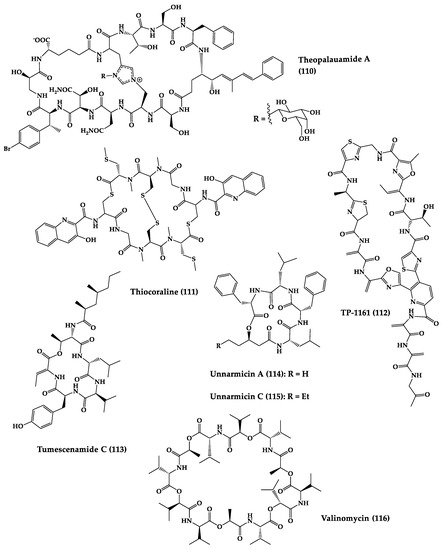
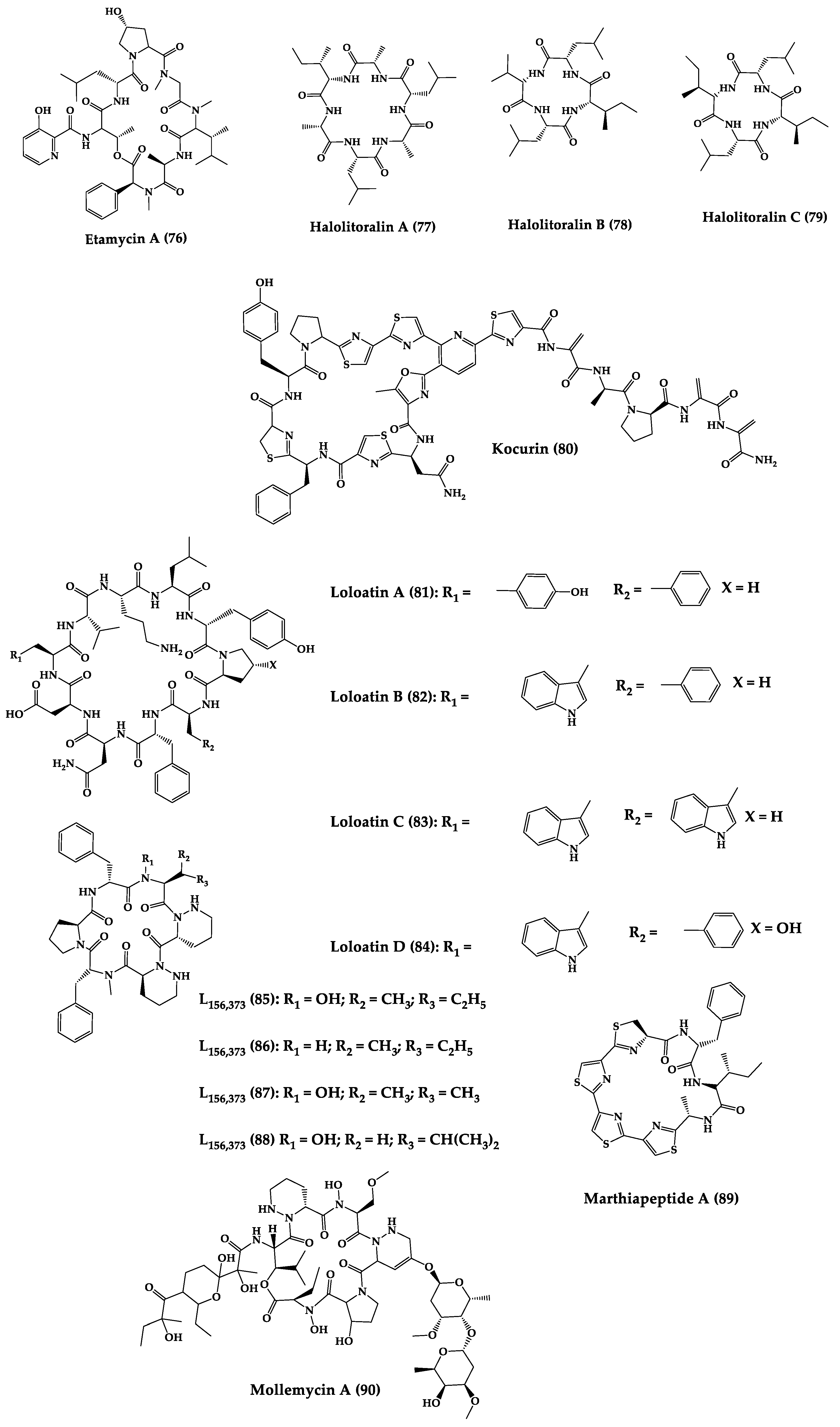
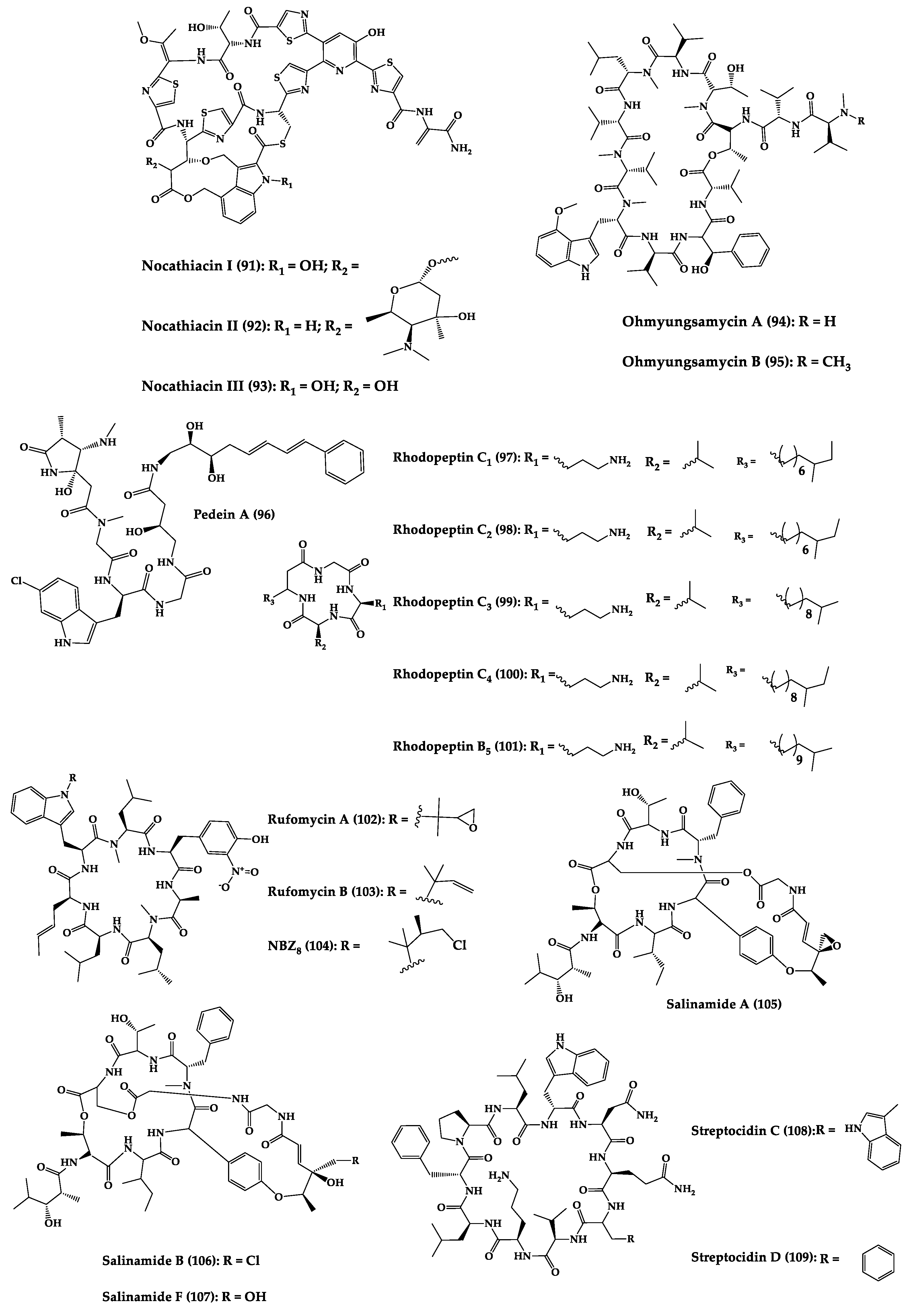
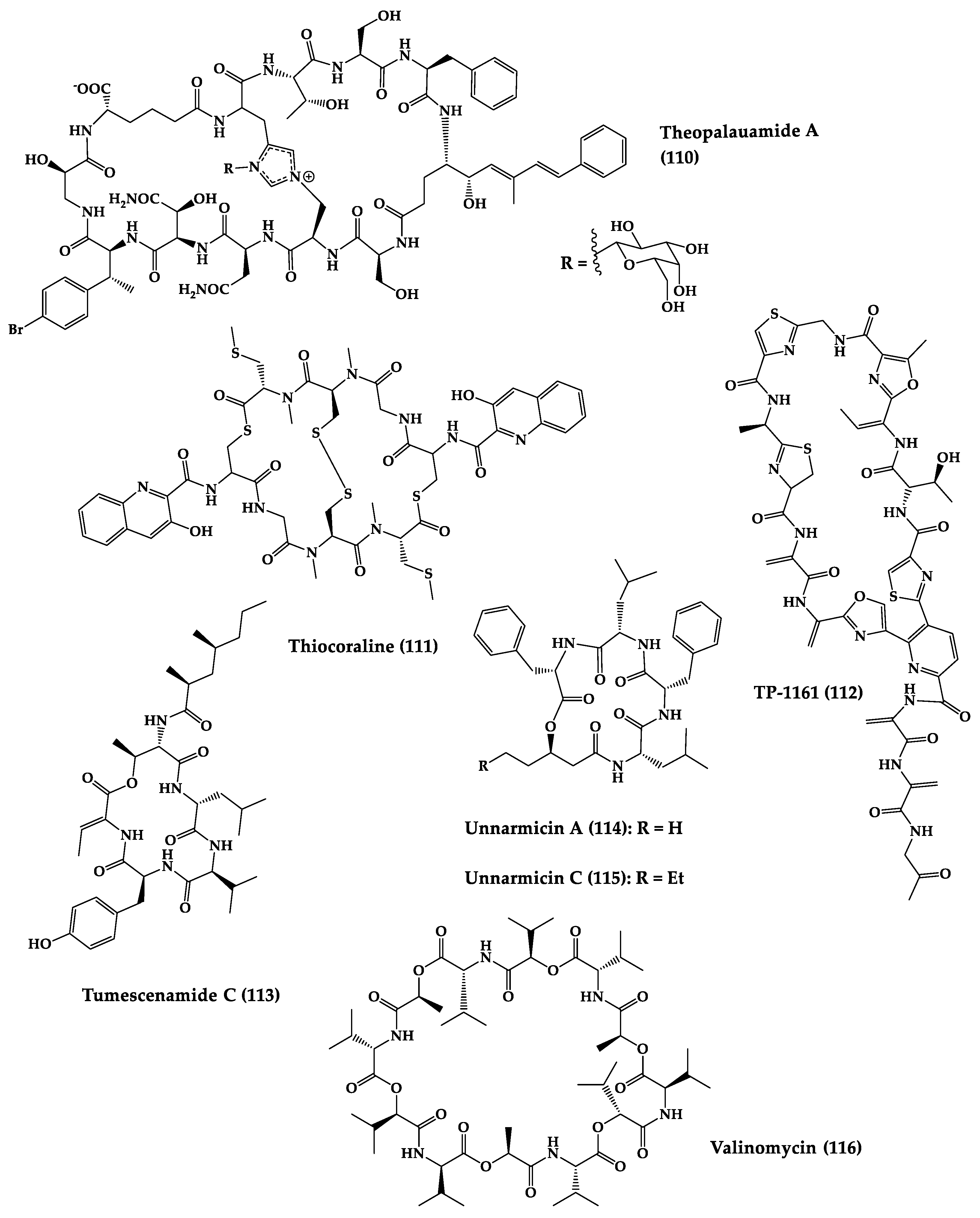
Figure 4.
Chemical structures of cyclic peptides from bacteria (
64
–
116
).