1. Obesity-Associated Modulation of Lung Immune Cells
A diverse population of immune cells residing in the lung plays a major role in defense surveillance. The aberrant activation of lung immune cells leads to the increased inflammation and remodeling of the airway, a commonly observed phenomenon in asthma
[1][46]. For instance, the activation of T helper cells (Th) residing in the lungs, such as Th2 and Th17, promotes the inflammation, mucous secretion, and remodeling of the airway in asthma via the secretion of different cytokines, such as IL-4, IL-5, IL-13, and IL-17, and interferon-γ (IFN-γ)
[2][47]. Alveolar macrophages are another type of lung immune cell that help in host defense by inducing inflammation via phagocytosis and the release of apoptotic bodies. Lung macrophages polarize into either the M1 phenotype during a non-allergic trigger or the M2 phenotype during allergic sensitization
[3][48]. The polarization of alveolar macrophages into the M2 phenotype is often observed in asthma
[4][5][49,50]. The activated alveolar macrophages are associated with defective phagocytosis, efferocytosis, altered remodeling and repairing of the airway, and increased activation of intracellular inflammasomes, thus indicating their role in asthma pathogenesis
[6][51]. In addition to these cells, lung immunity is also regulated by innate lymphoid cells (ILCs), a type of innate lymphocyte that synthesize various cytokines and promote inflammation and airway remodeling in asthma. For example, the induction of asthma phenotypes in mice by the administration of IL-25 or IL-33 through the intranasal route resulted in the accumulation of ILC2 in their BALF, lung, and mediastinal lymph nodes. Flow cytometry analysis showed that the accumulated ILC2 in lungs and BALF were associated with increased production of inflammatory cytokines, including IL-5 and IL-13. Similar outcomes were observed in house dust mite- or OVA-challenged asthmatic mice, implying that ILC2 plays a major role in inducing airway inflammation in asthma by enhancing the production of Th2 cytokines, such as IL-5 and IL-13
[7][52].
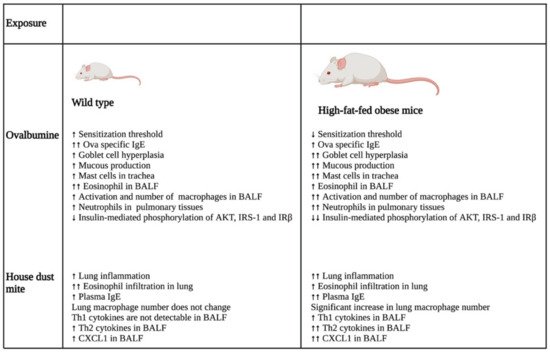
A study that investigated the impact of obesity on allergic inflammation in the airway indicated that obesity has an important role in modulating the immune response of T helper cells and macrophages (
Figure 12). OVA exposure results in increased expression of Th2 cytokines (IL-4, IL-5, IL-9, and IL-13) and Th17 cytokine, IL-17 A, in normal-weight BALB/c mice after 24 h, which was reduced after 48 h. On the other hand, high-fat-fed obese mice only had substantial increases in IL-9 and IL-13 after 24 h, which remained high even after 48 h of OVA challenge. Furthermore, these obese mice showed significant elevation of cytokines, such as IFN-γ, IL-4, IL-17A, TNF-α, IL-1β, and IL-6, after 48 h of OVA sensitization
[8][28].
Figure 12. Differential impacts on lung inflammation and remodeling after allergen exposure in normal and high-fat-fed in vivo mice models. In vivo studies indicated that normal diet-fed- and high-fat-fed mice had significant differences in inflammation, with the latter being associated with increased lung inflammation and remodeling, enhanced cytokine levels, and an elevated number of immune cells. ↑- increase; ↑↑- further increase due to additional effect; ↓- decrease; ↓↓- further decrease due to additional effect. Abbreviations: IgE: Immunoglobulin E; BALF: Bronchoalveolar lavage fluid; IRS-1: Insulin receptor substrate-1; IRβ: Insulin receptor β; CXCL1: Chemokine (C-X-C motif) ligand 1.
In the same study, the authors also showed that the OVA challenge resulted in the significant hyperplasia of goblet cells with increased mucous production in obese mice as compared with their normal-weight littermates. Furthermore, there was a substantial increase in the neutrophil population in the bone marrow of the obese group as compared with that of the lean group after 48 h of OVA sensitization. In addition, BALF analysis showed that the obese mice had a greater influx of macrophages in their BALF as compared with the lean mice after 24 and 48 h of OVA exposure. In contrast, eosinophil influx was lowered drastically in the BALF of obese mice as compared with that of the lean group at both time points. Furthermore, the macrophages in the allergic BALF of obese mice showed increased populations of arginase and inducible nitric oxide synthase-expressing macrophages as compared with those of non-obese, lean mice, indicating enhanced the activation of alveolar macrophages in the obese mice during allergic exposure. The pulmonary tissues of the obese mice had an increased number of neutrophils as compared with the lean mice after 48 h of OVA exposure. Collectively, the findings of the study imply that obesity impacts OVA-induced allergic inflammation in the airway of BALB/c mice by prolonging the inflammatory response of different Th cells, augmenting the influx of mixed granulocytes, and elevating macrophage activation and mucous production
[8][28].
Another study investigated the role of obesity-associated adipokine, leptin, in activating lung immune cells and inducing airway inflammation in asthma using Ob
−/− leptin-deficient mice
[9][27]. The study revealed that Ob
−/− mice showed reduced infiltration of lymphocytes and eosinophils in the BALFs and peribronchovascular spaces of their lungs compared with the wild-type littermates after the induction of experimental asthma by papain. Furthermore, inducing asthma by challenging with both papain and OVA resulted in reduced production of immunoglobulin (Ig) Es that are specific to OVA in the BALF and serum of Ob
−/− mice as compared with the control group, further indicating that leptin plays an essential role in inducing allergic airway inflammation in mice. Flow cytometry and enzyme-linked immunosorbent assay (ELISA) studies indicated that Ob
−/− asthmatic mice were associated with a reduced number of Th2 and ILC2 cells and decreased amounts of Th2 cytokines, including IL-4, IL-5, and IL-13, in their lung draining mediastinal lymph nodes as compared with the normal-weight asthmatic group. In complement to this finding, an in vitro study also indicated that exogenous leptin exposure promotes the expression of IL-4, IL-5, and IL-13 in Th2 cells without inducing their differentiation. Furthermore, leptin-deficient Ob
−/− asthmatic mice were also associated with fewer proliferating Th2 and ILC2 as compared with their normal-weight asthmatic littermates after the experimental induction of allergic asthma. In support of this, an in vitro study showed that leptin administration increases the proliferation of activated Th2 cells and promotes their survival. Since flow cytometry analysis showed the presence of leptin receptors on the surface of Th2 cells, the authors investigated the mechanism through which leptin induces their proliferation. Using specific inhibitors, the authors systematically demonstrated that leptin promotes the proliferation, survival, and cytokine production of Th2 cells by activating the PI3K-AKT-mTOR, JAK2-STAT3, and MAPK pathways
[9][27]. Collectively, the study indicated that the obesity-associated hormone leptin plays an indispensable role in promoting the inflammation of allergic airways in asthma by activating lung immune cells and modulating the influx of circulating immune cells.
2. Role of Obesity in Altering Airway Epithelium
The airway epithelium serves as a frontline barrier between the environment and the host system, and is exposed to infectious agents and airborne particles
[10][53]. Structurally, the epithelial layer is composed of ciliated columnar epithelial cells, intermediate columnal epithelial cells, goblet cells, basal cells, serous cells, and side population cells
[11][54]. The epithelial cells of the airway are endowed with different pattern recognition receptors, including Toll-like receptors, which are activated upon allergen and infectious agent exposure in asthmatic individuals. The activation of these receptors stimulates the secretion of various chemokines and cytokines from epithelial cells, such as IL-6, IL-8, IL-12, IL-12p40, IL-25, IL-33, CCL(C-C Motif chemokine ligand) 2, CCL20, GMCF (Granulocyte macrophage colony-stimulating factor), serum amyloid-A, and TSLP (Thymic stromal lymphopoietin), which subsequently triggers dendritic cell recruitment to the epithelial membrane
[12][13][14][15][55,56,57,58]. Recruited dendritic cells trigger the activation of innate and adaptive immunity, resulting in an inflammatory insult in the airway
[16][59]. Airway epithelial cells also release IL-33 that activates Th2 cells to secrete IL-5 and IL-13. These cytokines of the interleukin family promote the eosinophilic inflammation and metaplasia of goblet cells, resulting in airway inflammation and hyperresponsiveness
[17][60]. In addition, the airway epithelium also differentiates into a proliferative phenotype in asthma, leading to epithelial layer thickening in the bronchus
[18][61]. Activated airway epithelial cells can also augment airway remodeling by promoting the migration of airway smooth muscle (ASM) cells into the epithelial layer. For instance, an in vitro study showed that epithelial cells infected with human rhinovirus secrete various chemokines, such as CCL5, CXCL (Chemokine (C-X-C motif) ligand) 8, and CXCL10. Among these secreted chemokines, only CCL5 promoted the migration of ASMs
[19][62]. Taken together, these findings indicate that the epithelial cells of the airway play a pivotal role in inducing the inflammation and remodeling of the lung airway in asthma.
Recent studies have indicated that obesity induces the remodeling of airway epithelium via different mechanisms. A study by Elliot et al., 2019, showed that obese individuals are associated with excessive deposition of adipose tissue in the outer epithelial walls of medium and large airways that leads to airway wall thickening, consequently narrowing the airway
[20][63]. Another study by Suzukawa et al., 2015, investigated the molecular mechanism through which obesity modulates airway epithelial cell functioning. They found that the obesity hormone leptin increases the expression of the cell adhesion molecule, intercellular adhesion molecule (ICAM-1), in the nuclear factor kappa light-chain enhancer of activated B cells (NF-κB) in a dependent manner in human-derived primary bronchial epithelial cells and the BASB-2B airway epithelial cell line in vitro
[21][25]. ICAM-1 is a transmembrane glycoprotein that promotes the infiltration of eosinophils and neutrophils into the airway during inflammation
[22][23][64,65]. The study also showed that the leptin-mediated upregulation of ICAM-1 in BASB-2B cells was abrogated by dexamethasone treatment, indicating that BASB-2B cells are responsive to steroid treatment after leptin exposure. Using the multiplex cytokine analysis assay, the same study showed that leptin stimulation also increases the secretion of different cytokines, including IL-6, CCL11, Granulocyte-colony stimulating factor (G-CSF), and vascular endothelial growth factor (VEGF), in BSAB-2B cells. Flow cytometry analysis indicated that BEAS-2B cells express the leptin receptor Ob-R. Furthermore, transfecting these cells with Ob-R siRNA, followed by leptin treatment, did not show any upregulation of CCL11 mRNA, confirming that leptin modulates the phenotype of BEAS-2B cells by directly binding to its receptor. The study also demonstrated that leptin increased the proliferation and migration of airway epithelial cells and was able to protect TNF-α and IFN-γ-induced cell apoptosis in these cells
[21][25]. Taken together, the study indicated that leptin plays a key role in inducing the pro-inflammatory phenotype in airway epithelial cells. However, future investigations using in vivo models are necessary to confirm the role of leptin in inducing lung epithelial cell phenotypic switching in obesity-associated asthma.
Cysteinyl leukotrienes, a group of inflammatory lipid mediators, are found to be elevated in obese asthmatic individuals
[24][66]. A study by Dholia and coworkers investigated the role of leukotriene D4 (LTD4) in promoting the inflammation and remodeling of airway epithelial cells in vitro
[25][43]. LTD4 treatment increased the expression of inflammatory cytokines, such as IL-1α, IL-1β, IL-6, epidermal growth factor (EGF), TNF-α, granulocyte macrophage colony-stimulating factor (GM-CSF), and eotaxin, in small-airway epithelial cells (SAECs) in a time- and dose-dependent manner. The study also showed that LTD4 treatment induces the activation of the Natch domain-, leucine-rich repeat-, and PYD-containing protein 3 (NALP3) inflammasome by activating Caspase-1 and secreting IL-1β, indicating that LTD4 might serve as a danger molecule that binds to pattern recognition receptors on SAECs to induce inflammation. In support of this finding, LTD4 treatment also showed increased cyclooxygenase-2 (COX-2) inflammatory pathway activation in SAECs by enhancing the levels of COX-2 protein. In addition, using the air–liquid interface (ALI) technique, the authors of the study showed that LTD4 promotes the remodeling of SAECs by increasing the expression of vimentin and lowering the expression of E-cadherin, indicating the epithelial to mesenchymal transition of these cells. Furthermore, LTD4 treatment also increased the expression of Mucin5AC (Muc5AC), a marker of goblet cell hyperplasia, in SAECs grown in ALI culture. These molecular changes were associated with structural alterations in SAECs, such as the loss of cilia and excessive accumulation of mucin. Further investigation by the same group to understand the molecular mechanism involved in LTD4-mediated remodeling of SAECs indicated that LTD4 treatment increases the expression of transforming growth factor (TGF)-β that consequently phosphorylates Smad2/3 and activates the TGF-β/Smad2/3 pathway
[25][43].
3. Obesity-Induced Alteration of Airway Smooth Muscle Cell Phenotype
Airway smooth muscle cells form another important structural component of the airway that plays an important role in regulating the structure, function, and bronchomotor tone of the bronchial airway. In asthma, the smooth muscle cells of the bronchus induce the contraction of the airway by altering the intracellular Ca
2+ levels
[26][67]. Furthermore, the ASMs of asthmatic individuals show an increased size (hypertrophy), elevated proliferation rate (hyperplasia), and enhanced migration to the airway epithelial layer, leading to the increased thickness of the airway in asthma
[27][68]. The thickness of the ASM area has been found to be positively linked to asthma severity independent of the disease period, indicating that ASM layer thickening is an early event in asthma that determines the severity of the disease
[28][69]. Airway smooth muscle cells can also induce a proinflammatory environment in asthma by secreting a myriad of inflammatory cytokines, including a family of interleukins (IL), such as IL-1, IL-5, IL-6, and IL-8, and growth factors, including TGF-β1 and VEGF
[29][30][70,71]. Thus, differentiated airway smooth muscle cells play a critical role in the inflammation, narrowing, and remodeling of the bronchial airway in asthma.
The increased activation of airway smooth muscle cells in response to various external and internal cues results in their hyper contraction by enhancing the release of calcium from the sarcoplasmic reticulum and subsequent phosphorylation of the myosin light chain. These events consequently lead to airway hyperresponsiveness in asthma
[31][32][33][72,73,74]. Obesity is shown to aggravate airway hyperresponsiveness in asthmatic individuals to a higher extent. A study by Orfanos et al., 2018, investigated the impact of obesity on the airway smooth muscle cell response to contractile antagonists, carbachol, and histamine
[34][30]. The results of their study showed that carbachol stimulation induces the enhanced phosphorylation of the myosin light chain in the human airway smooth muscle (HASM) cells of obese individuals as compared with those from age- and sex-matched lean individuals. Using single-cell calcium analysis, the authors of the study informed that both carbachol and histamine provoke intracellular calcium mobilization in HASM cell lines derived from obese subjects to a significantly higher extent than those originating from normal-weight donors. Among obese individuals, the HASM cells of women had higher intracellular calcium release in response to carbachol as compared with those from men. The HASM cells of obese females also showed increased intracellular calcium release as compared with those from comparable nonobese female counterparts; however, no significant difference was observed between the HASM cells of obese and nonobese males. Of note, the intracellular calcium response of HASM cells derived from obese donors was comparable to that of those obtained from fatal asthma patients, indicating that obese individuals can develop airway hyperresponsiveness to an extent similar to asthma patients. In support of these findings, a fluorescently labeled elastomeric contractible surfaces (FLECS) assay showed that the HASM cells of patients with obesity undergo greater shortening after carbachol and histamine exposure as compared with those of nonobese individuals. Taken together, the findings of this study indicate that obesity might play a dominant role in augmenting airway hyperresponsiveness in asthma by modulating the mechanism of cell contraction in airway smooth muscle cells
[34][30].
Increased levels of free fatty acids (FFAs) are frequently reported in obese individuals
[35][75]. Elevated levels of FFAs are known to induce the phenotypic switching of various types of cells, such as airway smooth muscle cells, by binding to their endogenous receptors on the cells’ surface. A systematic study by Matoba et al., 2017, showed that long-chain fatty acids, such as oleic acid and linoleic acid, and GW9508, an agonist of FFA receptor 1 (FFAR1), induce the proliferation of HASM cells in vitro by activating the MEK/ERK and PI3K/AKT pathways
[36][76]. Mechanistically, long-chain FFAs and GW9508 activate MEK/ERK and PI3K/AKT signaling cascades by phosphorylating ERK and AKT in vitro in HSAM cell lines and patient-derived HASM cells, as well as in airway smooth muscle isolated from rats ex vivo. Further investigations from the same group also indicated that one of the long-chain FFAs, oleic acid, induces the phosphorylation of ERK and AKT in HASM cells by binding to the FFAR1 receptor that associates with G proteins, Gα
i and Gα
q, on the cell membrane, leading to the dissociation of Gβγ subunits from Gα proteins. Furthermore, the study showed that FFAR1-coupled Gα
i and Gα
q, and dissociated Gβγ subunits activate the c-Raf/ERK pathway, leading to ERK phosphorylation, whereas Gβγ
i induces the phosphorylation of AKT by activating ras and Src without involving Gβγ subunits. The study also revealed that the phosphorylation of AKT and ERK proceeds through PI3K and MEK, respectively, further leading to the phosphorylation of p70S6K, which eventually phosphorylates the S6 ribosomal protein. These events consequently promote airway smooth muscle cell proliferation.
4. Obesity and Airway Fibroblasts
Lung fibroblasts play a vital role in asthma pathogenesis by promoting airway remodeling and fibrosis by secreting different inflammatory cytokines and extracellular matrix proteins
[37][38][77,78]. A recent investigation indicated that leptin exposure enhances the production of various cytokines and chemokines, such as eotaxin, monocyte chemoattractant protein-1 (MCP-1), IL-6, IL-8, and interferon gamma-induced protein 10 (IP-10), in normal lung fibroblasts. However, the siRNA-mediated knockdown of Ob-R receptors in these cells suppresses the leptin-mediated upregulation of IL-6, CCL11, and IP-10, indicating that leptin can play a detrimental role in promoting inflammation in asthma patients by stimulating fibroblast differentiation
[39][31]. However, further investigations are needed to understand the other mechanisms through which obesity promotes fibrosis in asthma.