Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Muhammad Ikram Ullah and Version 2 by Peter Tang.
Oculocutaneous albinism (OCA) is an autosomal recessive syndromic and non-syndromic defect with deficient or a complete lack of the melanin pigment. The characteristics of OCA appears in skin, hair, and eyes with variable degree of pigmentation. Clinical manifestations of OCA include nystagmus, photophobia, reduced visual acuity, hypo-plastic macula, and iris trans-illumination. There are eight OCA types (OCA1–8) documented with non-syndromic characteristics.
- albinism
- consanguinity
- non-syndromic oculocutaneous albinism
1. OCA Albinism
OCA is a genetic disorder with an autosomal recessive mode of inheritance in which there is a complete absence or decreased production of melanin that causes hypo-pigmentation of hair, skin, and eyes. Each parent transfers a copy of the faulty gene to the affected child in OCA. Physically normal carriers of OCA can transfer the disease to the next generation due to the presence of only one copy of the defective gene or compound heterozygous mutations in two different alleles of OCA genes, which have also been identified in different populations of the world, establishing the heterogeneity of this disorder [1][7].
Albinism is also present in syndromic form with variable signs and symptoms such as neurological problems, with high susceptible to infections [2][8]. Hermansky–Pudlak, Chediak–Higashi, Griscelli Syndrome, Prader–Willi, Angelman, Cross–McKusick–Breen, Griscelli, Elejalde, and Waardenburg Syndrome type II (WS2) are the main examples of syndromic albinism. All of these syndromes except WS2, are autosomal recessive and can be diagnosed by clinical, biochemical, and genetic testing.
On the basis of genetic testing, non-syndromic OCA is further divided into eight subtypes (OCA1–8), among which OCA1 is the most common that affects almost 50% of albino individuals worldwide [3][9].
2. Melanin Biosynthesis Pathway and Its Link to OCA Types
In melanosomes, the molecular mechanism regulates the synthesis of melanin pigment by melanocyte cells. In this pathway, there are a number of genes and contributing enzymes that take part in multiple reactions to make melanin products. In a chemical reaction, L-tyrosine is converted into a 3,4-dihydroxyphenylalanine (DOPA) intermediate that then results in the formation of dopaquinone. From dopaquinone, the melanin synthetic pathway splits into two pathways leading to eumelanin formation (brownish to black coloration) and pheomelanin (reddish to yellow pigmentation) ultimately (Figure 1). Previous studies documented the crucial role of the genes (TYR, OCA2, TYRP1, SLC45A2, SLC24A5, C10orf11, and DCT) in modulating the regulation of melanin production [4][5][10,11]. The initiation of the pathway is regulated by the tyrosinase enzyme that is a membrane-bound copper-containing enzyme that causes the hydroxylation of l-tyrosine into L-DOPA, and then dopaquinone is produced. Apart from tyrosinase a, some proteins present in melanosomes, including tyrosinase-related protein types 1 and 2 (TYRP1, TYRP2), have key functions in eumelanin production. The role of TYRP1 is to increase the ratio of eumelanin: pheomelanin production, while it also plays its role as an anti-oxidant due to the presence of peroxidase activity [6][7][12,13]. A number of transcription factors, genes, and hormones including stem cell factor (SCF), SOX10, PAX3, WNT, MITF, ACTH, and α-MSH are involved in the melanin synthesis regulating pathway. It has been proposed that more than 125 genes contribute to its functional activity in melanogenesis. The defects in structural proteins, enzymes, ligands, hormones, and receptors of melanin pigment production result in loss of functions in these factors including TYR, OCA2, TYRP1, TYRP2, SLC24A5, DCT, MATP, and MC1R, which contribute to phenotypes of subtypes of OCA from OCA1–8 with an autosomal recessive non-syndromic pattern [7][8][13,14].
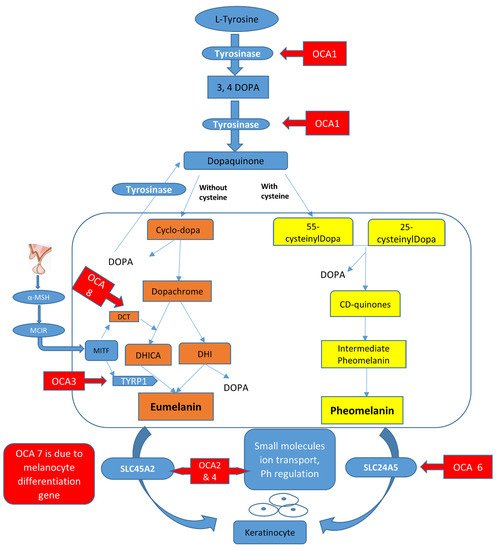
Figure 1.
Graphical representation of the melanin synthesis pathway and rate-limiting steps that if defective lead to OCA types.
3. Prevalence/Epidemiology
Overall, the prevalence of OCA is projected at a frequency of 1 in 17,000–20,000. The mutant allele of OCA2 is carried in approximately 1 in 70 individuals, categorized as the prevalent type of albinism worldwide, although the frequency of mutation detection is higher in the OCA1 phenotype. The frequency of OCA2 alleles is more prevalent in the sub-Saharan African population, with a rate of 1 in 1000 cases [9][10][15,16]. This is due to the cultural customs that allow marriages within blood relations, a phenomenon is known as pseudo-dominance. In different populations, the prevalence of the specific types of albinism vary. The prevalence of type 1 of OCA (almost 1 in 40,000 worldwide) is the most common type (70% of cases) in America, China (sporadic albinism), the subcontinent (mostly familial albinism), and in the Caucasian population [11][12][13][14][17,18,19,20]. The frequency of OCA2 is the most common type with a prevalence rate of 1 in 39,000 worldwide. This type of albinism is more frequent in the population in sub-Saharan Africa, with an approximate rate of 1 in 3900, followed by African–Americans and Americans [9][10][15,16]. These two types of OCA are also seen in Pakistani and Indian families with frequent genetic mutations [3][11][15][9,17,21]. The prevalence of OCA type 3 and type 4 is 1 in 8500 and 1 in 100,000, respectively. OCA3 is reported in German, Japanese, and Indo–Pakistani populations. OCA4 is frequent in the Japanese population with a rate of 24% of overall OCA followed by different countries in Asia and Europe. OCA type 5 is only reported in a consanguineous Pakistani family [16][22], while cases of OCA6 and OCA7 are reported in China, eastern India, and Atlantic Island [17][18][19][23,24,25], but the exact prevalence has not been documented. Syndromic OCA is also variable in prevalence. The prevalence of Hermansky–Pudlak syndrome (HPS) is 1 in approximately 500,000 worldwide, the rate of Chediak–Higashi syndrome (CHS) is not evidently reported (almost 500 cases reported), and the prevalence rate for Angelman syndrome (AS) and Prader–Willi syndrome (PWS) is 1 in 12,000 and 1 in 15,000, respectively, i.e., a higher frequency rate than OCA types. The latter types of syndromic albinism are presented, such as OCA2, and 1% of cases showed gene deletions [20][26].
4. Phenotype Variations in OCA
Clinically, there are seven OCA forms that present a wide range of phenotypes, signs, and symptoms. OCA1 is the frequent type that is further divided into OCA1A and OCA1B subtypes [21][22][23][24][27,28,29,30]. OCA1A has a severe phenotype due to the complete absence of melanin pigment that leads to discoloration of skin and hair with a transparent iris in affected cases. All other forms of OCA including OCA1B and OCA2–OCA8 may present some coloration of the tissues such as hair, skin, and eyes with age development. The variety of pigmentation varies, displaying the extent of colors, but absolute pigment production is never achieved [25][31]. The photosensitivity of skin is also improved due to the absence of photo-protective melanin pigment in the skin. In OCA types, clinical presentations linked to eyes develop due to misrouting of the optic nerve and are presented as photophobia, congenital nystagmus, translucence hypo-pigmented iris, refractive errors, pigment scarcity in the epithelium, occasionally color vision impairment and foveal hypoplasia, reduction of visual acuity (VA) usually between 20/60 to 20/400, strabismus, and reduced stereoscopic vision due to misrouting of the optic nerve, which are the important OCA phenotype characteristics [26][27][28][32,33,34].
During the embryonic development stage, the melanin deficiency leads to eye developmental defects such as the retina and iris lacking melanin pigment, foveal hypoplasia, misrouting of optic nerve fibers of the visual cortex, and small or elongated globes [26][32]. As a result of melanin absence, a slight color is imparted to the eyes in albinism cases. Normal eye functioning is dependent on melanin pigment; therefore, impaired vision occurs due to low or deficient pigment availability. The main reason for impaired or loss of vision is under-development of the macula in the retina, leading to foveal hypoplasia. The other functions of melanin are to contribute to hair and skin coloration. The reduced production of melanin causes the development of a variety of colors in hair such as white to red or golden, light yellow or very light blonde [28][34]. In skin, the deficiency of pigment produces a milky-white and deep fair color in some cases. The complications of albinism result in severe damage of the skin from sunburn and may progress to skin cancers in later stages [28][34].