Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 3 by Conner Chen.
The proline-catalyzed asymmetric Michael addition reaction of acetaldehyde with α,β-unsaturated nitroalkenes as synthetically useful routes to β-substituted derivatives of γ-Aminobutyric acid (GABA).
- pharmaceuticals
- neurological drugs
- γ-aminobutyric-acid derivatives
- asymmetric Michael addition
1. Introduction
Tailor-made amino acids [1] play an indispensable role in the development of modern pharmaceuticals and drug formulations [2][3][4][5][6]. Thus, over 20% of newly approved small-molecule drugs contain structural fragments of AAs [7][8][9]. In particular, γ-aminobutyric-acid (GABA) derivatives bearing β-alkyl or β-aryl substituents, which include baclofen [10], phenibut [11][12][13], tolibut [14], and pregabalin [15][16][17], are recognized as an essential class of marketed pharmaceuticals for the treatment of neurological diseases (Figure 1). Similarly, piracetam-based GABA derivatives such as phenylpiracetam [18], brivaracetam [19][20][21], and rolipram [22] are also developed as pharmaceuticals. Introduction of β-alkyl or β-aryl substituent in the GABA backbone allows for the improvement of the lipophilic character of these compounds.
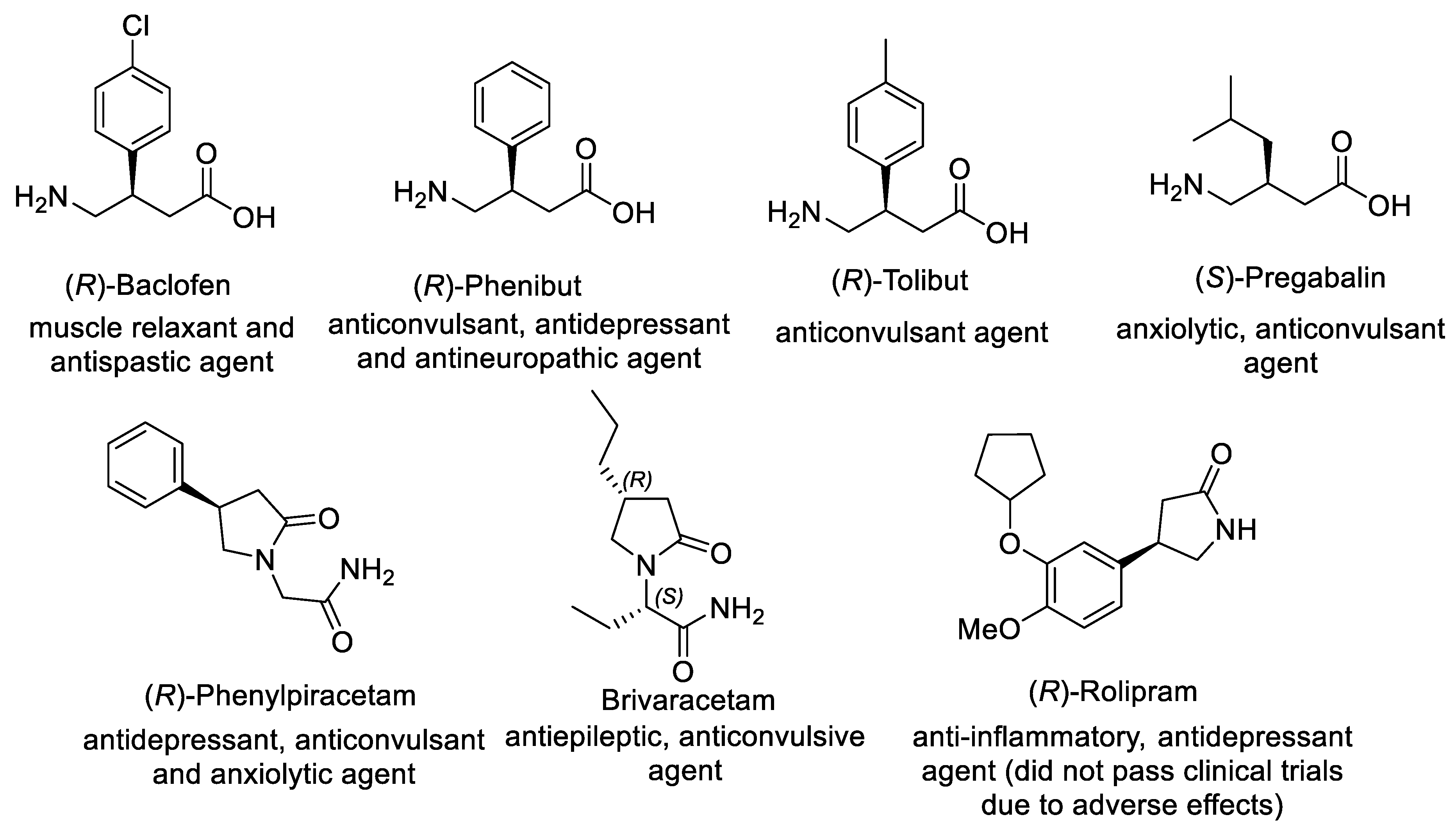
Figure 1. Representative β-substituted GABA derivatives with clinical applications.
Significantly, biological activity of β-substituted GABA derivatives depends on their absolute configuration. For example, (R)-enantiomers of Baclofen (antispastic agent and muscle relaxant) and Phenibut (tranquilizer and anticonvulsant) are considerably more active than the corresponding (S)-enantiomers, while anticonvulsant activity of Pregabalin (anti-epilepsy drug) is primarily related to (S)-enantiomer [23]. Consequently, considerable efforts were devoted to developing asymmetric synthesis of β-substituted GABA derivatives, including chemical and biocatalytic resolution, asymmetric reduction, desymmetrization, aldol addition, and nucleophilic substitution [24][25][26]. For the past decade, the asymmetric Michael addition of carbon nucleophile to α,β-unsaturated compound-bearing nitro or carbonyl groups gained impressive progress, providing straightforward access to γ-nitrocarbonyl compounds as key chiral intermediates that can be converted into β-substituted GABA derivatives via subsequent transformation of functional groups.
2. Michael Addition of Carbonyl Compounds to α,β-Unsaturated Nitroalkenes
The proline-catalyzed asymmetric Michael addition reaction [27] of acetaldehyde with α,β-unsaturated nitroalkenes attracted considerable attention as synthetically useful routes to β-substituted derivatives of GABA. Thus, the addition of acetaldehyde to the nitroolefins (E)-1 and (E)-2 (Scheme 1) was carried out in the presence of enantiomerically pure (S)-diphenylprolinol silyl ether 3 as the catalyst [28][29]. After optimization of reaction conditions, the asymmetric Michael addition proceeded efficiently with 10–20 mol% of organocatalyst (S)-3 in such solvents as MeCN, DMF/i-PrOH, and 1,4-dioxane to afford γ-nitro aldehydes (S)-4 and (R)-5 in reasonable yield and excellent enantiomeric excess. Oxidation of γ-nitro aldehydes (S)-4 and (R)-5 was successfully performed in aqueous t-BuOH using NaClO2 and NaH2PO4 with 2-methyl-2-butene as a chlorine scavenger to afford carboxylic acids (S)-6 and (R)-7 in good to excellent yields [30]. The reduction of the nitro acid (S)-6 with Raney Ni in MeOH gave, after treatment with aqueous HCl, (S)-Baclofen 8 as hydrochloride salt in 91% yield. (R)-Pregabalin 9 was also synthesized by the reduction of the nitro group in (R)-7 under Pd/C in 93% yield. The mechanism of the asymmetric Michael addition reaction of acetaldehyde with nitroalkenes promoted by diphenylprolinol silyl ether involves the formation of the enamine as a nucleophile. Thus, the organocatalyst would react with the acetaldehyde forming anti-enamine with the double bond oriented away from the diphenylsiloxymethyl group. In this case, the (diphenylmethyl)trimethylsiloxy group provides the formation of anti-enamine and shielding one face of the enamine double bond. The anti-enamine would add stereoselectively to the nitroolefin via the acyclic synclinal transition state proposed by Seebach [31] as shown in Scheme 1.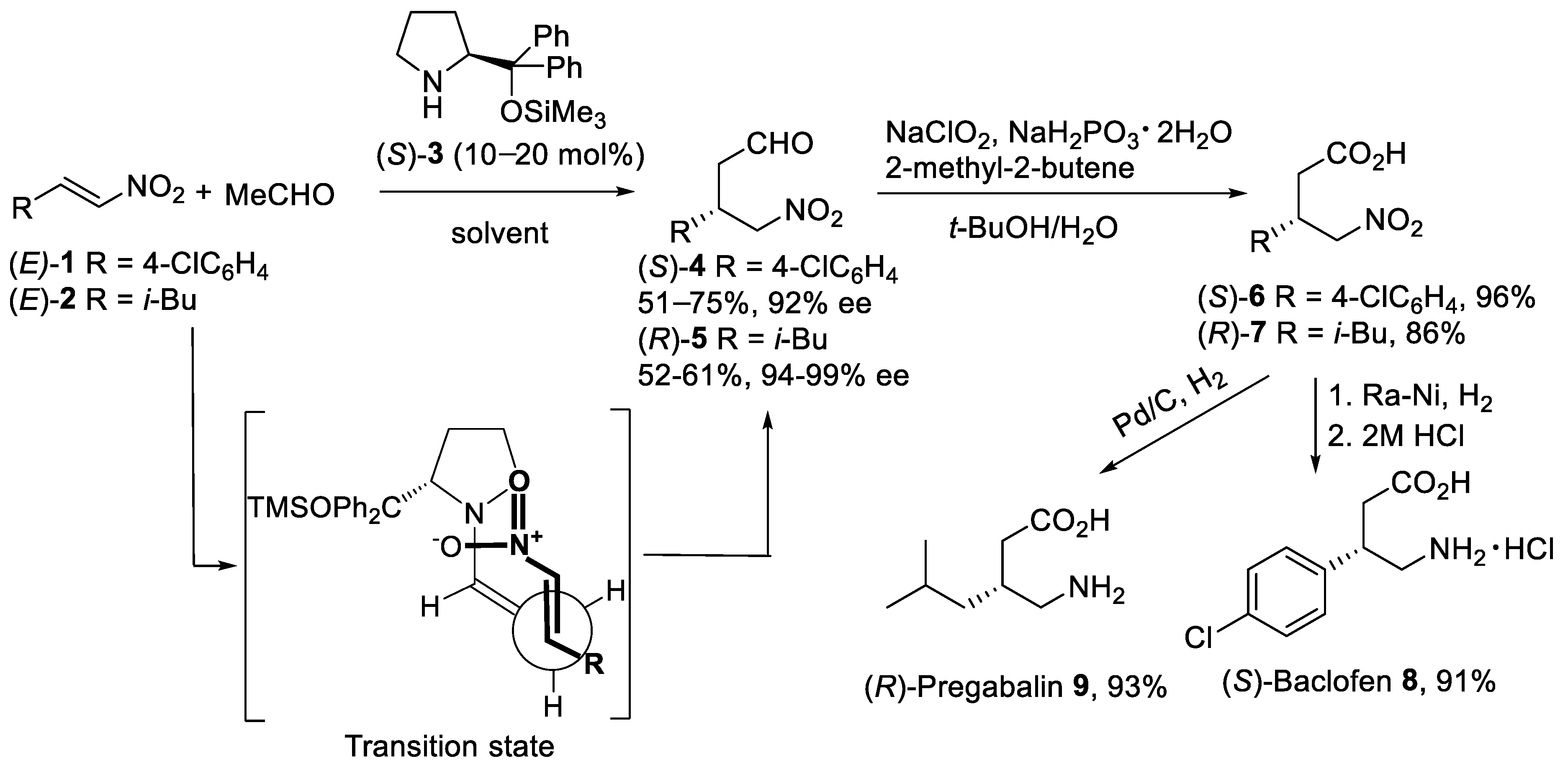
Scheme 1. Organocatalytic asymmetric Michael reaction of acetaldehyde with nitroalkenes (E)-1 and (E)-2.
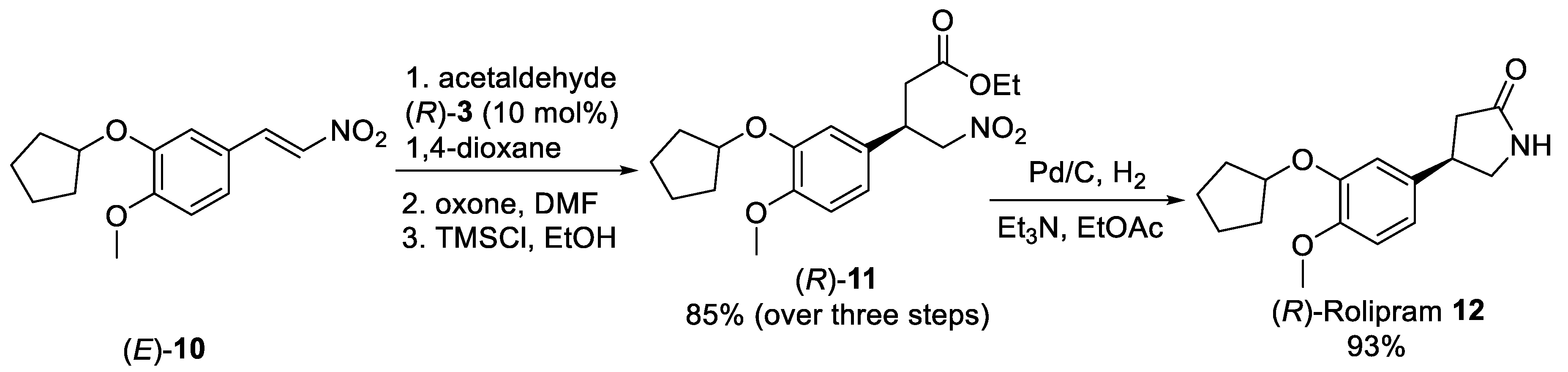
Scheme 2. Synthetic approach of (R)-Rolipram 12 employing the organocatalyzed asymmetric Michael addition acetaldehyde to nitro olefin (E)-10.
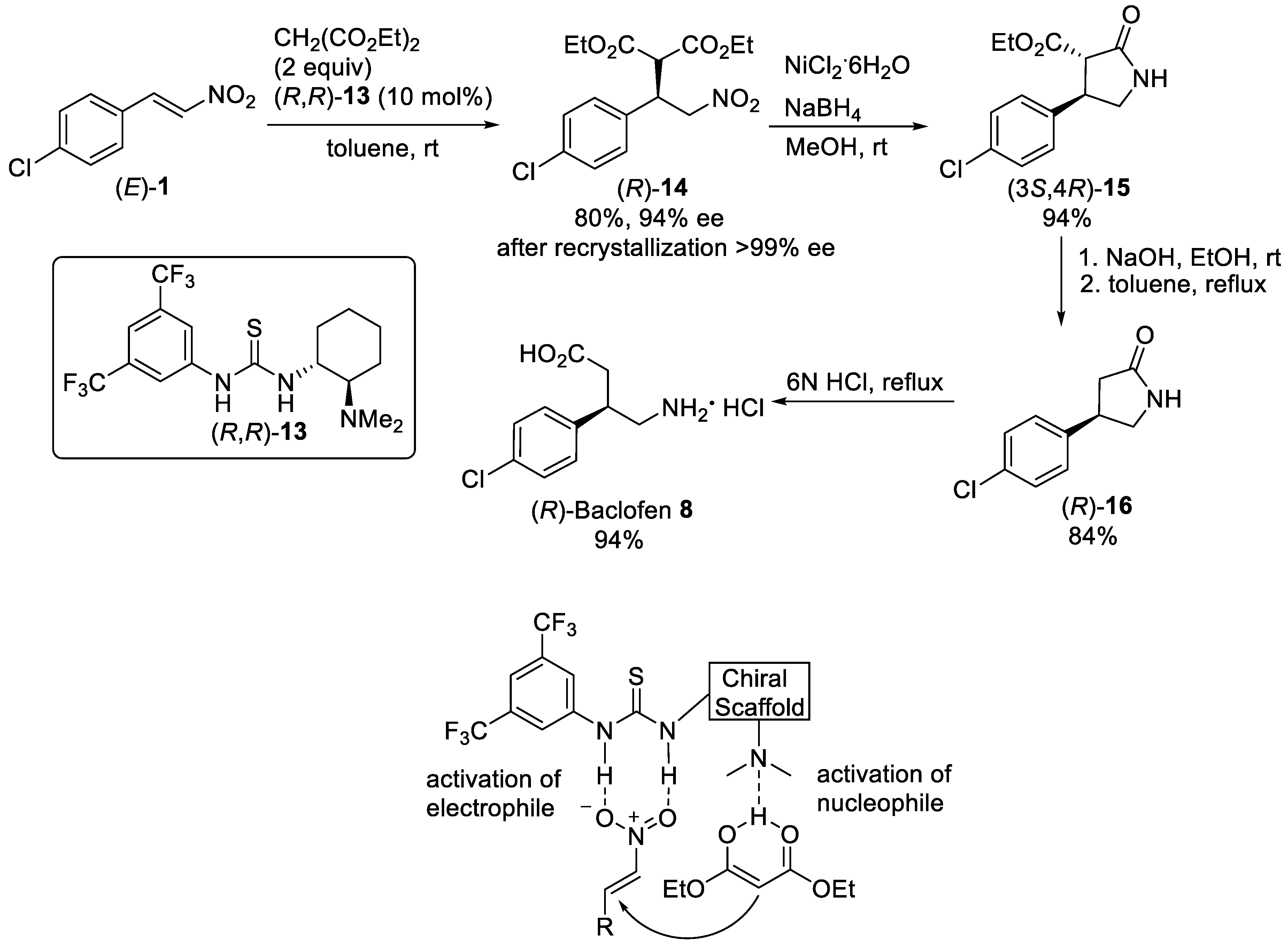
Scheme 3. Enantioselective Michael reaction of (E)-1 with diethyl malonate in the presence of thiourea (R,R)-13.
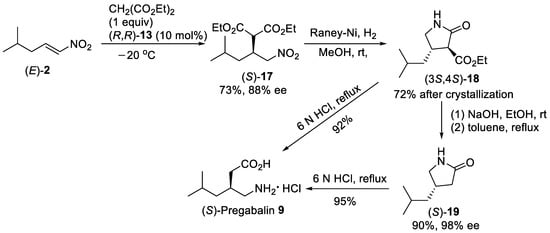
Scheme 4. Enantioselective Michael reaction of (E)-2 with diethyl malonate in the presence of thiourea (R,R)-13 under solvent-free conditions.
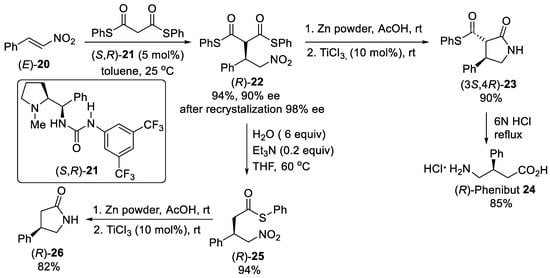
Scheme 5. Enantioselective Michael addition of diphenyl dithiomalonates to β-nitrostyrene (E)-20 catalyzed by L-proline-derived urea (S,R)-21.
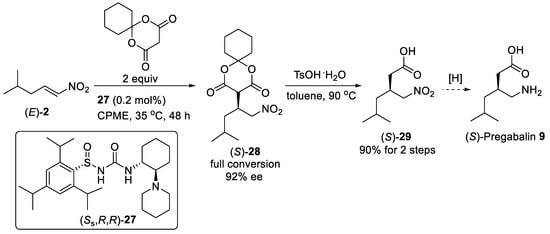
Scheme 6. Catalytic enantioselective addition of cyclohexyl Meldrum’s acid to nitroalkene (E)-2 using N-sulfinyl urea catalysis (Ss,R,R)-27.
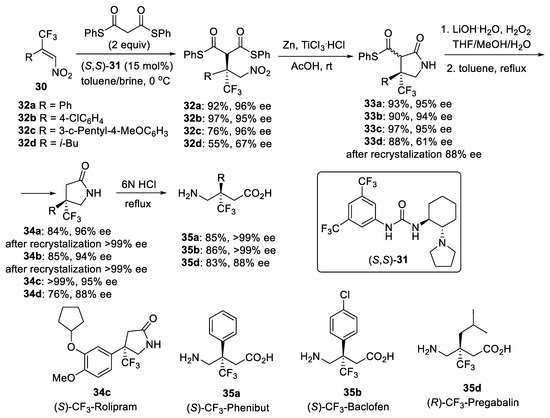
Scheme 7. Synthesis of GABA derivatives with a trifluoromethylated quaternary stereogenic center.
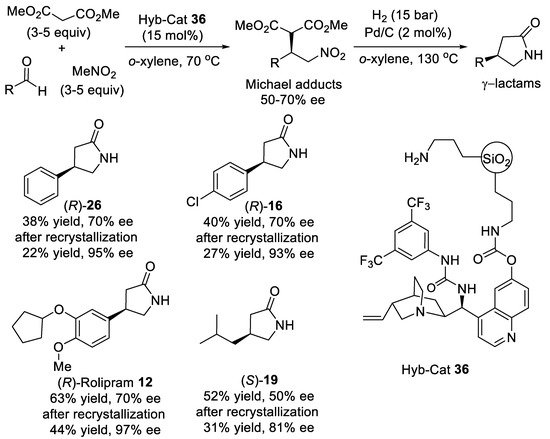
Scheme 8. Asymmetric three-component reaction of aldehydes, nitromethane, and malonate.
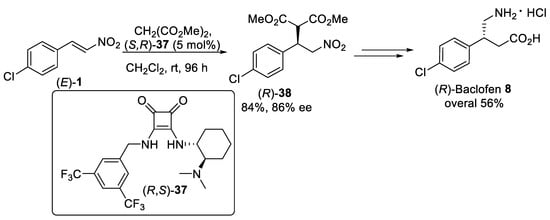
Scheme 9. Michael addition of dimethyl malonate to β-nitrostyrene (E)-1 using squaramide organocatalysis (R,S)-37.
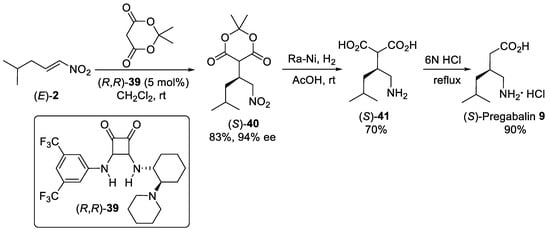
Scheme 10. The Michael addition of Meldrum’s acid to nitroalkene (E)-2 catalyzed by squaramide (R,R)-39 catalyst.
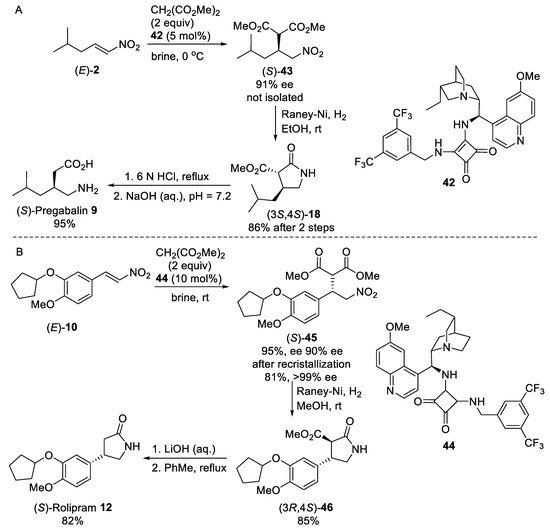
Scheme 11. Enantioselective syntheses of (S)-Pregabalin 9 (A) and (S)-Rolipram 12 (B) under “on water” conditions.
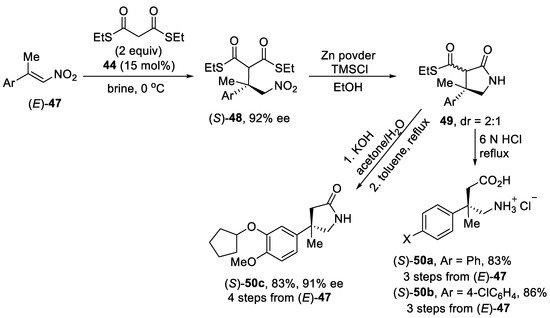
Scheme 12. Enantioselective Michael addition reaction of β,β-disubstituted nitroalkenes (E)-47 with dithiomalonate using dihydroquinine-squaramide catalyst.
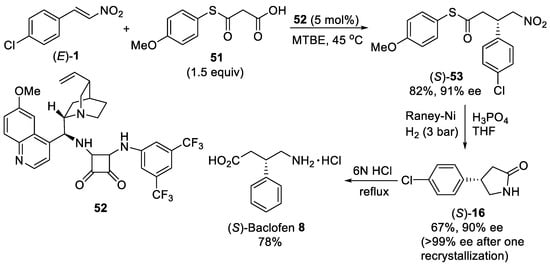
Scheme 13. Enantioselective Michael addition of malonic-acid half-thioester 51 to β-nitrostyrene (E)-1. Synthesis of (S)-baclofen 8.
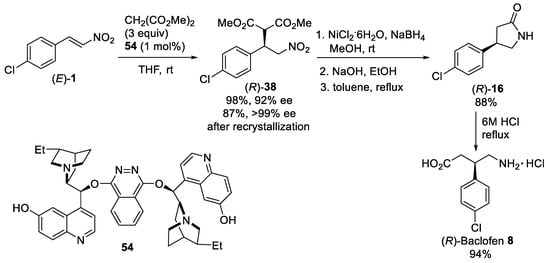
Scheme 14. Asymmetric conjugate addition of dimethyl malonate to β-nitrostyrene (E)-1. Synthesis of (R)-Baclofen 8 hydrochloride.
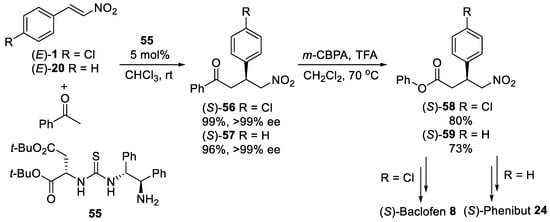
Scheme 15. Michael reaction between acetophenone and β-nitrostyrenes using primary amine-thiourea organocatalyst 55.
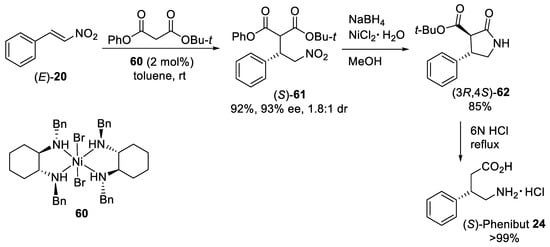
Scheme 16. Enantioselective Michael additions of tert-butyl phenyl malonate to β-nitrostyrene (E)-20 catalyzed by diamine−Ni(II) complex 60.
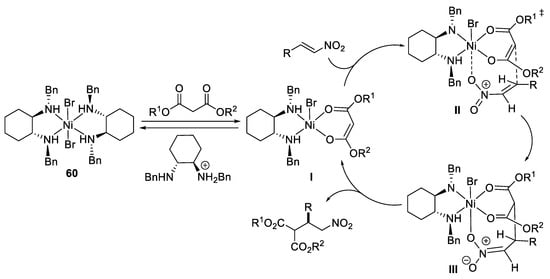
Scheme 17. Proposed mechanism reaction.
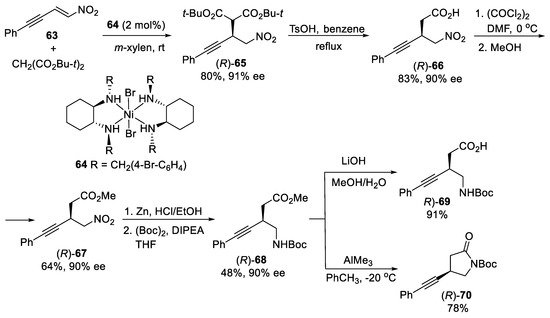
Scheme 18. Asymmetric synthesis of β-alkynyl-γ-amino acid (R)-69 and β-alkyny-γ-lactam (R)-70.
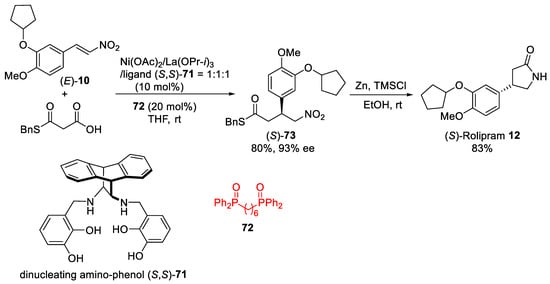
Scheme 19. Ni–La system for decarboxylative 1,4-addition of malonic-acid half-thioester to nitroalkene (E)-10.
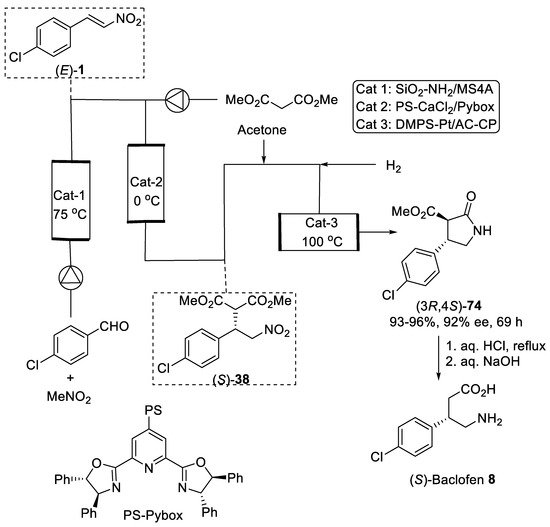
Figure 2. Sequential flow synthesis of Baclofen precursor (3R,4S)-74.
3. Michael Additions of Cyanide or Nitroalkanes to α,β-Unsaturated Carbonyl Compounds
Asymmetric conjugate addition of cyanide and nitroalkanes to α,β-unsaturated carbonyl substrates was another practical route for the synthesis of β-substituted GABA derivatives. For example, the Michael addition of diethylaluminum cyanide to substrate (R)-75 (Scheme 20) bearing oxazolidinone chiral auxiliary [78] was conducted as the key step in the synthesis of (S)-Pregabalin 9 from commercially available starting materials [79]. Conjugate addition was performed in toluene at 0 °C to produce addition adduct (4R,3′S)-76 in satisfactory yield and moderate dr (87:13). The diastereomerically pure (4R,3′S)-76 was provided after purification with column chromatography on silica gel in a 57% yield. The observed stereoselectivity was attributed to the approach of the diethylaluminum cyanide mainly from the less hindered Si face opposite to phenyl group in the oxazolidinone auxiliary. The removal of oxazolidinone chiral auxiliary by treating with LiOH and H2O2 in aqueous THF and reduction of the cyano group by hydrogenolysis under Raney Ni afforded (S)-pregabalin 9 in 95% yield for two steps. Moreover, acetone cyanohydrin was also effectively used as cyanide source for diastereoselective conjugate addition to α,β-unsaturated oxazolidinone (S)-77 (Scheme 20) [80]. The hydrocyanation of (S)-77 cleanly proceeded with two equivalents of acetone cyanohydrin and 10 mol% of Sm(III) isopropoxide as catalyst in toluene to give the addition adduct (4S,3′R)-78 in 75% yield and 88:12 dr. The diastereomerically pure product (4S,3′R)-78 was chromatographically isolated in 66% yield. The catalytic hydrogenation of the cyano group with simultaneous cleavage of oxazolidinone chiral auxiliary over platinum oxide afforded the lactam (R)-79 in 75% yield and 96% ee, which acidic hydrolysis led to the (R)-Pregabalin 9 as hydrochloride in 95% yield with retention of the enantiomeric purity. A similar route was employed for the synthesis of (S)-Baclofen 8 using aryl-substituted substrate (S)-80, acetone cyanohydrin and Sm(Oi-Pr)3 as catalyst (Scheme 20) [80]. Under standard conditions, the diastereomerically pure nitrile adduct (4S,3′S)-81 was obtained in 62% yield. Selective reduction of the cyano group with such reagents as NaBH4 and NiCl2 and hydrolysis of resulting lactam (S)-82 provided (S)-Baclofen 8 in excellent yield and enantiomeric purity.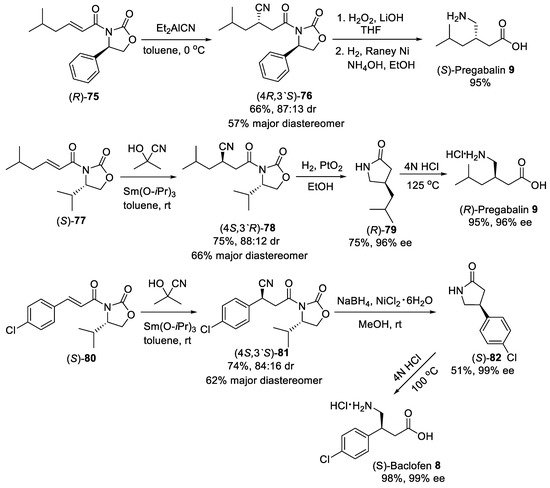
Scheme 20. Diastereoselective Michael addition of cyanide to α,β-unsaturated oxazolidinones.
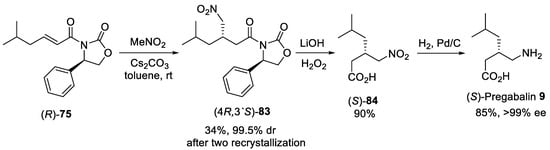
Scheme 21. Preparation of (S)-Pregabalin 9.
References
- Soloshonok, V.A.; Cai, C.; Hruby, V.J.; Van Meervelt, L. Asymmetric synthesis of novel highly sterically constrained (2S, 3S)-3-methyl-3-trifluoromethyl-and (2S, 3S, 4R)-3-trifluoromethyl-4-methylpyroglutamic acids. Tetrahedron 1999, 55, 12045–12058.
- Han, J.; Lyutenko, N.V.; Sorochinsky, A.E.; Okawara, A.; Konno, H.; White, S.; Soloshonok, V.A. Tailor-Made Amino Acids in Pharmaceutical Industry: Synthetic Approaches to Aza-Tryptophan Derivatives. Chem. Eur. J. 2021, 27, 17510–17528.
- Han, J.; Konno, H.; Sato, T.; Soloshonok, V.A.; Izawa, K. Tailor-made amino acids in the design of small-molecule blockbuster drugs. Eur. J. Med. Chem. 2021, 220, 113448.
- Han, J.; Konno, H.; Sato, T.; Izawa, K.; Soloshonok, V.A. Peptidomimetics and Peptide-Based Blockbuster Drugs. Curr. Org. Chem. 2021, 25, 1627–1658.
- Liu, J.; Han, J.; Izawa, K.; Sato, T.; White, S.; Meanwell, N.A.; Soloshonok, V.A. Cyclic tailor-made amino acids in the design of modern pharmaceuticals. Eur. J. Med. Chem. 2020, 208, 112736.
- Mei, H.; Han, J.; White, S.; Graham, D.J.; Izawa, K.; Sato, T.; Fustero, S.; Meanwell, N.A.; Soloshonok, V.A. Tailor-made amino acids and fluorinated motifs as prominent traits in modern pharmaceuticals. Chem. Eur. J. 2020, 26, 11349–11390.
- Liu, A.; Han, J.; Nakano, A.; Konno, H.; Moriwaki, H.; Abe, H.; Izawa, K.; Soloshonok, V.A. New pharmaceuticals approved by FDA in 2020: Small-molecule drugs derived from amino acids and related compounds. Chirality 2022, 34, 86–103.
- Yin, Z.; Hu, W.; Zhang, W.; Konno, H.; Moriwaki, H.; Izawa, K.; Han, J.; Soloshonok, V.A. Tailor-made amino acid-derived pharmaceuticals approved by the FDA in 2019. Amino Acids 2020, 52, 1227–1261.
- Soloshonok, V.A.; Izawa, K. (Eds.) Asymmetric Synthesis and Application of α-Amino Acids; ACS Symposium Series #1009; Oxford University Press: Washington, DC, USA, 2009.
- Kent, C.N.; Park, C.; Lindsley, C.W. Classics in chemical neuroscience: Baclofen. ACS Chem. Neurosci. 2020, 11, 1740–1755.
- Lapin, I. Phenibut (β-phenyl-GABA): A tranquilizer and nootropic drug. CNS Drug Rev. 2001, 7, 471–481.
- Zvejniece, L.; Vavers, E.; Svalbe, B.; Veinberg, G.; Rizhanova, K.; Liepins, V.; Kalvinsh, I.; Dambrova, M. R-phenibut binds to the α2–δ subunit of voltage-dependent calcium channels and exerts gabapentin-like anti-nociceptive effects. Pharmacol. Biochem. Behav. 2015, 137, 23–29.
- Dambrova, M.; Zvejniece, L.; Liepinsh, E.; Cirule, H.; Zharkova, O.; Veinberg, G.; Kalvinsh, I. Comparative pharmacological activity of optical isomers of phenibut. Eur. J. Pharmacol. 2008, 583, 128–134.
- Tyurenkov, I.N.; Borodkina, L.E.; Bagmetova, V.V.; Berestovitskaya, V.M.; Vasil’eva, O.S. Comparison of nootropic and neuroprotective features of aryl-substituted analogs of gamma-aminobutyric acid. Bull. Exp. Biol. Med. 2016, 160, 465–469.
- Silverman, R.B. From basic science to blockbuster drug: The discovery of Lyrica. Angew. Chem. Int. Ed. 2008, 47, 3500–3504.
- Reinares, M.; Rosa, A.R.; Franco, C.; Goikolea, J.M.; Fountoulakis, K.; Siamouli, M.; Gonda, X.; Frangou, S.; Vieta, E. A systematic review on the role of anticonvulsants in the treatment of acute bipolar depression. Int. J. Neuropsychop. 2013, 16, 485.
- Calandre, E.P.; Rico-Villademoros, F.; Slim, M. Alpha2delta ligands, gabapentin, pregabalin and mirogabalin: A review of their clinical pharmacology and therapeutic use. Expert Rev. Neurother. 2016, 16, 1263–1277.
- Zvejniece, L.; Zvejniece, B.; Videja, M.; Stelfa, G.; Vavers, E.; Grinberga, S.; Svalbe, B.; Dambrova, M. Neuroprotective and anti-inflammatory activity of DAT inhibitor R-phenylpiracetam in experimental models of inflammation in male mice. Infammopharmacology 2020, 28, 1283–1292.
- Malykh, A.G.; Sadaie, M.R. Piracetam and piracetam-like drugs. Drugs 2010, 70, 287–312.
- Kenda, B.M.; Matagne, A.C.; Talaga, P.E.; Pasau, P.M.; Differding, E.; Lallemand, B.I.; Frycia, A.M.; Moureau, F.G.; Klitgaard, H.V.; Gillard, M.R.; et al. Discovery of 4-substituted pyrrolidone butanamides as new agents with significant antiepileptic activity. J. Med. Chem. 2004, 47, 530–549.
- von Rosenstiel, P. Brivaracetam (ucb 34714). Neurotherapeutics 2007, 4, 84–87.
- Zhu, J.; Mix, E.; Winblad, B. The antidepressant and antiinflammatory effects of rolipram in the central nervous system. CNS Drug Rev. 2001, 7, 387–398.
- Gajcy, K.; Lochyński, S.; Librowski, T. A role of GABA analogues in the treatment of neurological diseases. Curr. Med. Chem. 2010, 17, 2338–2347.
- Ordóñez, M.; Cativiela, C.; Romero-Estudillo, I. An update on the stereoselective synthesis of γ-amino acids. Tetrahedron Asymmetry 2016, 27, 999–1055.
- Ramesh, P.; Suman, D.; Reddy, K.S.N. Asymmetric Synthetic Strategies of (R)-(–)-Baclofen: An Antispastic Drug. Synthesis 2018, 50, 211–226.
- Martínez, C.A.; Hu, S.; Dumond, Y.; Tao, J.; Kelleher, P.; Tully, L. Development of a chemoenzymatic manufacturing process for pregabalin. Org. Process Res. Dev. 2008, 12, 392–398.
- Shagufta, K.; Ameer, F.Z.; Sajjad, A.; Rabia, A.; Iqra, K.; Wajiha, Q.; Attia, M. Recent trends in the development of novel catalysts for asymmetric Michael reaction. Curr. Org. Chem. 2020, 24, 1397–1458.
- García-García, P.; Ladépêche, A.; Halder, R.; List, B. Catalytic asymmetric Michael reactions of acetaldehyde. Angew. Chem. Int. Ed. 2008, 47, 4719–4721.
- Hayashi, Y.; Itoh, T.; Ohkubo, M.; Ishikawa, H. Asymmetric Michael reaction of acetaldehyde catalyzed by diphenylprolinol silyl ether. Angew. Chem. Int. Ed. 2008, 47, 4722–4724.
- Gotoh, H.; Ishikawa, H.; Hayashi, Y. Diphenylprolinol silyl ether as catalyst of an asymmetric, catalytic, and direct Michael reaction of nitroalkanes with α, β-unsaturated aldehydes. Org. Lett. 2007, 9, 5307–5309.
- Seebach, D.; Golinski, J. Synthesis of Open-Chain 2,3-Disubstituted 4-nitroketones by Diastereoselective Michael-addition of (E)-Enamines to (E)-Nitroolefins. A topological rule for C, C-bond forming processes between prochiral centres. Preliminary communication. Helv. Chim. Acta 1981, 64, 1413–1423.
- Kaur, R.; Pandey, S.K. Efficient synthesis of (−)-(R)-and (+)-(S)-rolipram. Tetrahedron Lett. 2017, 58, 4333–4335.
- Qiao, Y.; He, J.; Ni, B.; Headley, A.D. Asymmetric Michael reaction of acetaldehyde with nitroolefins catalyzed by highly water-compatible organocatalysts in aqueous media. Adv. Synth. Catal. 2012, 354, 2849–2853.
- Nori, V.; Sinibaldi, A.; Giorgianni, G.; Pesciaioli, F.; Di Donato, F.; Cocco, E.; Biancolillo, A.; Landa, A.; Carlone, A. DoE-Driven Development of an Organocatalytic Enantioselective Addition of Acetaldehyde to Nitrostyrenes in Water. Chem. Eur. J. 2022, 28, e202104524.
- Palomo, C.; Landa, A.; Mielgo, A.; Oiarbide, M.; Puente, A.; Vera, S. Water-compatible iminium activation: Organocatalytic Michael reactions of carbon-centered nucleophiles with enals. Angew. Chem. Int. Ed. 2007, 46, 8431–8435.
- Alonso, D.A.; Baeza, A.; Chinchilla, R.; Gómez, C.; Guillena, G.; Pastor, I.M.; Ramón, D.R. Recent advances in asymmetric organocatalyzed conjugate additions to nitroalkenes. Molecules 2017, 22, 895.
- Odagi, M.; Nagasawa, K. Recent Advances in Natural Products Synthesis Using Bifunctional Organocatalysts bearing a Hydrogen-Bonding Donor Moiety. Asian J. Org. Chem. 2019, 8, 1766–1774.
- Okino, T.; Hoashi, Y.; Furukawa, T.; Xu, X.; Takemoto, Y. Enantio-and diastereoselective Michael reaction of 1, 3-dicarbonyl compounds to nitroolefins catalyzed by a bifunctional thiourea. J. Am. Chem. Soc. 2005, 127, 119–125.
- Perlikowska, R.; Piekielna, J.; Mazur, M.; Koralewski, R.; Olczak, J.; do Rego, J.C.; Fichna, J.; Modranka, J.; Janecki, T.; Janecka, A. Antinociceptive and antidepressant-like action of endomorphin-2 analogs with proline surrogates in position 2. Bioorg. Med. Chem. 2014, 22, 4803–4809.
- Sukhanova, A.A.; Nelyubina, Y.V.; Zlotin, S.G. Asymmetric synthesis of 3-prenyl-substituted pyrrolidin-2-ones. Mendeleev Commun. 2016, 26, 471–473.
- Steppeler, F.; Dominika Iwan, D.; Elżbieta Wojaczyńska, E.; Wojaczyński, J. Chiral thioureas—Preparation and significance in asymmetric synthesis and medicinal chemistry. Molecules 2020, 25, 401.
- Liu, J.; Wang, X.; Ge, Z.; Sun, Q.; Cheng, T.; Li, R. Solvent-free organocatalytic Michael addition of diethyl malonate to nitroalkenes: The practical synthesis of Pregabalin and γ-nitrobutyric acid derivatives. Tetrahedron 2011, 67, 636–640.
- Bae, H.Y.; Song, C.E. Unprecedented Hydrophobic Amplification in Noncovalent Organocatalysis “on Water”: Hydrophobic Chiral Squaramide Catalyzed Michael Addition of Malonates to Nitroalkenes. ACS Catal. 2015, 5, 3613–3619.
- Jin, H.; Kim, S.T.; Hwang, G.S.; Ryu, D.H. l-Proline Derived Bifunctional Organocatalysts: Enantioselective Michael Addition of Dithiomalonates to trans-β-Nitroolefins. J. Org. Chem. 2016, 81, 3263–3274.
- Kimmel, K.L.; Weaver, J.D.; Ellman, J.A. Enantio-and diastereoselective addition of cyclohexyl Meldrum’s acid to β-and α, β-disubstituted nitroalkenes via N-sulfinyl urea catalysis. Chem. Sci. 2012, 3, 121–125.
- Mondal, A.; Bhowmick, S.; Ghosh, A.; Chanda, T.; Bhowmick, K.C. Advances on asymmetric organocatalytic 1,4-conjugate addition reactions in aqueous and semi-aqueous media. Tetrahedron Asymmetry 2017, 28, 849–875.
- Sim, J.H.; Park, J.H.; Maity, P.; Song, C.E. Access to Chiral GABA Analogues Bearing a Trifluoromethylated All-Carbon Quaternary Stereogenic Center through Water-Promoted Organocatalytic Michael Reactions. Org. Lett. 2019, 21, 6715–6719.
- Leyva-Pérez, A.; García-García, P.; Corma, A. Multisite organic–inorganic hybrid catalysts for the direct sustainable synthesis of GABAergic drugs. Angew. Chem. Int. Ed. 2014, 53, 8687–8690.
- García-García, P.; Zagdoun, A.; Copèret, C.; Lesage, A.; Díaz, U.; Corma, A. In situ preparation of a multifunctional chiral hybrid organic–inorganic catalyst for asymmetric multicomponent reactions. Chem. Sci. 2013, 4, 2006–2012.
- Veverková, E.; Bilka, S.; Baran, R.; Šebesta, R. Squaramide-Catalyzed Michael Addition as a Key Step for the Direct Synthesis of GABAergic Drugs. Synthesis 2016, 48, 1474–1482.
- Marchetti, L.A.; Kumawat, L.K.; Mao, N.; Stephens, J.C.; Elmes, R.B.P. The versatility of squaramides: From supramolecular chemistry to chemical biology. Chem 2019, 5, 1398–1485.
- Baran, R.; Veverková, E.; Škvorcová, A.; Šebesta, R. Enantioselective Michael addition of 1, 3-dicarbonyl compounds to a nitroalkene catalyzed by chiral squaramides—A key step in the synthesis of pregabalin. Org. Biomol. Chem. 2013, 11, 7705–7711.
- Bassas, O.; Huuskonen, J.; Rissanen, K.; Koskinen, A.M.P. A simple organocatalytic enantioselective synthesis of pregabalin. Eur. J. Org. Chem. 2009, 2009, 1340–1351.
- Kitanosono, T.; Kobayashi, S. Synthetic Organic “Aquachemistry” that Relies on Neither Cosolvents nor Surfactants. ACS Cent. Sci. 2021, 7, 739–747.
- Bae, H.Y.; Some, S.; Oh, J.S.; Lee, Y.S.; Song, C.E. Hydrogen bonding mediated enantioselective organocatalysis in brine: Significant rate acceleration and enhanced stereoselectivity in enantioselective Michael addition reactions of 1,3-dicarbonyls to β-nitroolefins. Chem. Commun. 2011, 47, 9621–9623.
- Sim, J.H.; Song, C.E. Water-Enabled Catalytic Asymmetric Michael Reactions of Unreactive Nitroalkenes: One-Pot Synthesis of Chiral GABA-Analogs with All-Carbon Quaternary Stereogenic Centers. Angew. Chem. Int. Ed. 2017, 56, 1835–1839.
- Wang, Z.L. Recent advances in catalytic asymmetric decarboxylative addition reactions. Adv. Synth. Catal. 2013, 355, 2745–2755.
- Bae, H.Y.; Some, S.; Lee, J.H.; Kim, J.Y.; Song, M.J.; Lee, S.; Zhang, Y.J.; Song, C.E. Organocatalytic Enantioselective Michael-Addition of Malonic Acid Half-Thioesters to β-Nitroolefins: From Mimicry of Polyketide Synthases to Scalable Synthesis of γ-Amino Acids. Adv. Synth. Catal. 2011, 353, 3196–3202.
- Lubkoll, J.; Wennemers, H. Mimicry of Polyketide Synthases—Enantioselective 1, 4-Addition Reactions of Malonic Acid Half-Thioesters to Nitroolefins. Angew. Chem. Int. Ed. 2007, 46, 6841–6844.
- Li, F.; Li, Y.Z.; Jia, Z.S.; Xu, M.H.; Tian, P.; Lin, G.Q. Biscinchona alkaloids as highly efficient bifunctional organocatalysts for the asymmetric conjugate addition of malonates to nitroalkenes at ambient temperature. Tetrahedron 2011, 67, 10186–10194.
- Serdyuk, O.V.; Heckel, C.M.; Tsogoeva, S.B. Bifunctional primary amine-thioureas in asymmetric organocatalysis. Org. Biomol. Chem. 2013, 11, 7051–7071.
- Tsakos, M.; Kokotos, C.G.; Kokotos, G. Primary Amine-Thioureas with Improved Catalytic Properties for “Difficult” Michael Reactions: Efficient Organocatalytic Syntheses of (S)-Baclofen, (R)-Baclofen and (S)-Phenibut. Adv. Synth. Catal. 2012, 354, 740–746.
- Zheng, K.; Liu, X.; Feng, X. Recent advances in metal-catalyzed asymmetric 1,4-conjugate addition (ACA) of nonorganometallic nucleophiles. Chem. Rev. 2018, 118, 7586–7656.
- Hui, C.; Pu, F.; Xu, J. Metal-Catalyzed Asymmetric Michael Addition in Natural Product Synthesis. Chem. Eur. J. 2017, 23, 4023–4036.
- Evans, D.A.; Mito, S.; Seidel, D. Scope and mechanism of enantioselective Michael additions of 1,3-dicarbonyl compounds to nitroalkenes catalyzed by nickel (II)–diamine complexes. J. Am. Chem. Soc. 2007, 129, 11583–11592.
- Liu, K.; Jin, R.; Ceng, T.; Xu, X.; Gao, F.; Liu, G.; Li, H. Functionalized Periodic Mesoporous Organosilica: A Highly Enantioselective Catalyst for the Michael Addition of 1,3-Dicarbonyl Compounds to Nitroalkenes. Chem. Eur. J. 2012, 18, 15546–15553.
- Bissessar, D.; Achard, T.; Bellemin-Laponnaz, S. Robust and Recyclable Self-Supported Chiral Nickel Catalyst for the Enantioselective Michael Addition. Adv. Synth. Catal. 2016, 358, 1982–1988.
- Poe, S.L.; Kobašlija, M.; McQuade, D.T. Mechanism and application of a microcapsule enabled multicatalyst reaction. J. Am. Chem. Soc. 2007, 129, 9216–9221.
- Ishitani, H.; Kanai, K.; Yoo, W.J.; Yoshida, T.; Kobayashi, S. A Nickel-Diamine/Mesoporous Silica Composite as a Heterogeneous Chiral Catalyst for Asymmetric 1, 4-Addition Reactions. Angew. Chem. Int. Ed. 2019, 58, 13313–13317.
- Buendia, M.B.; Kegnæs, S.; Kramera, S. A Nickel-Bisdiamine Porous Organic Polymer as Heterogeneous Chiral Catalyst for Asymmetric Michael Addition to Aliphatic Nitroalkenes. Adv. Synth. Catal. 2020, 362, 5506–5512.
- Li, X.; Li, X.; Peng, F.; Shao, Z. Mutually Complementary Metal- and Organocatalysis with Collective Synthesis: Asymmetric Conjugate Addition of 1,3-Carbonyl Compounds to Nitroenynes and Further Reactions of the Products. Adv. Synth. Catal. 2012, 354, 2873–2885.
- Matsunaga, S.; Shibasaki, M. Recent advances in cooperative bimetallic asymmetric catalysis: Dinuclear Schiff base complexes. Chem. Commun. 2014, 50, 1044–1057.
- Furutachi, M.; Mouri, S.; Matsunaga, S.; Shibasaki, M. A Heterobimetallic Ni/La-salan Complex for Catalytic Asymmetric Decarboxylative 1,4-Addition of Malonic Acid Half-Thioester. Chem. Asian J. 2010, 5, 2351–2354.
- Tsubogo, T.; Yamashita, Y.; Kobayashi, S. Toward efficient asymmetric carbon–carbon bond formation: Continuous flow with chiral heterogeneous catalysts. Chem. Eur. J. 2012, 18, 13624–13628.
- Ishitani, H.; Furiya, Y.; Kobayashi, S. Enantioselective Sequential-Flow Synthesis of Baclofen Precursor via Asymmetric 1,4-Addition and Chemoselective Hydrogenation on Platinum/Carbon/Calcium Phosphate Composites. Chem. Asian. J. 2020, 15, 1688–1691.
- Ishitani, H.; Saito, Y.; Tsubogo, T.; Kobayashi, S. Synthesis of Nitro-Containing Compounds through Multistep Continuous Flow with Heterogeneous Catalysts. Org. Lett. 2016, 18, 1346–1349.
- Tsubogo, T.; Oyamada, H.; Kobayashi, S. Multistep continuous-flow synthesis of (R)-and (S)-rolipram using heterogeneous catalysts. Nature 2015, 520, 329–332.
- Zadsirjan, V.; Heravi, M.M. Oxazolidinones as Chiral Auxiliaries in the Asymmetric 1,4-Conjugate Addition Reaction Applied to the Total Synthesis of Natural Products: A Supplemental Mini-Review. Curr. Org. Synth. 2018, 15, 3–20.
- Tovar-Gudiño, E.; Morales-Nava, R.; Fernández-Zertuche, M. Diasteroselective conjugate addition of diethylaluminum cyanide to a conjugated N-enoyl system: An alternative synthesis of (S)-pregabalin. Can. J. Chem. 2014, 92, 45–48.
- Armstrong, A.; Convine, N.J.; Popkin, M.E. Diastereoselective conjugate addition of cyanide to α, β-unsaturated oxazolidinones: Enantioselective synthesis of ent-pregabalin and baclofen. Synlett 2006, 2006, 1589–1591.
- He, C.; Zhai, Z.; Zhou, Y.; Li, J.; Wang, G. A new synthetic route for the preparation of pregabalin. Synth. Commun. 2021, 51, 2034–2040.
More