Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Zuzana Országhová and Version 3 by Jessie Wu.
Testicular germ cell tumors (GCTs) are highly curable malignancies. Excellent survival rates in patients with metastatic disease can be attributed to the exceptional sensitivity of GCTs to cisplatin-based chemotherapy. This hypersensitivity is probably related to alterations in the DNA repair of cisplatin-induced DNA damage, and an excessive apoptotic response. However, chemotherapy fails due to the development of cisplatin resistance in a proportion of patients, who are then considered “platinum-refractory”.
- germ cell tumors
- cisplatin
- chemoresistance
- mechanisms
1. Introduction
The molecular basis of cisplatin resistance in germ cell tumors (GCTs) appears to be multifactorial. The mechanisms of cisplatin resistance in general have been classified by some authors according to the sequence of processes that follow the introduction of the drug into the human body: pre-target, on-target and post-target mechanisms. Pre-target resistance includes alterations occurring before cisplatin binds to DNA in the cell, on-target resistance refers to alterations directly related to DNA-cisplatin adducts, and post-target resistance involves alterations in downstream signaling pathways of cisplatin-mediated DNA damage that lead to apoptosis [1][2][3][4]. Although the mechanisms within cancer cells are still the main subject of research, increasing evidence has demonstrated that the tumor microenvironment and immune cells are also important in the development of cisplatin resistance [3]. The possible mechanisms involved in cisplatin resistance are summarized in Figure 1.
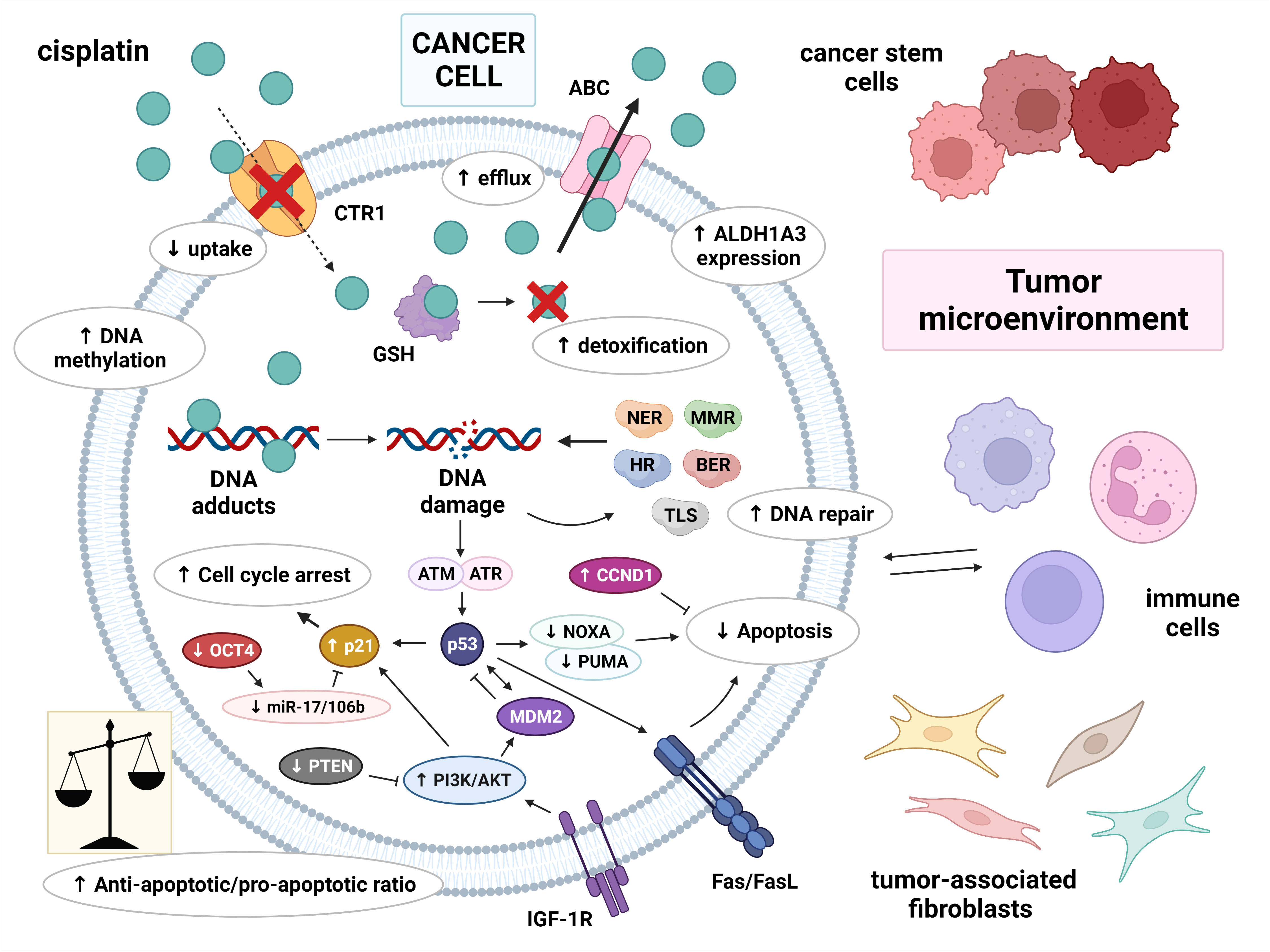
Figure 1. Multiple molecular mechanisms are responsible for cisplatin resistance in germ cell tumors. Cisplatin resistance can be classified as pre-target, on-target and post-target. Pre-target resistance includes: decreased intracellular accumulation of cisplatin (reduced uptake and increased efflux of cisplatin) and increased cisplatin detoxification by cytoplasmic scavengers. On-target resistance refers to an increased ability to repair DNA damage or an acquired ability to tolerate unrepaired DNA lesions. Post-target resistance involves: alterations in apoptosis signaling pathways with a central role of p53, decreased expression of pro-apoptotic factors, and overexpression of anti-apoptotic factors. This leads to cell cycle arrest and the inhibition of apoptosis. Other factors that contribute to cisplatin resistance are: DNA hypermethylation, increased ALDH expression, and a tumor microenvironment with immune cells playing an important role. Abbreviations: ABC = ATP-binding cassette transporter; AKT = protein kinase B (PKB); ALDH1A3 = aldehyde dehydrogenase 1A3; ATM = ataxia telangiectasia mutated; ATR = ATM and RAD3-related; BER = base excision repair; CCND1 = cyclin D1; CTR1 = copper transporter protein; FasL = Fas ligand; GSH = glutathione; HR = homologous recombination; IGF-1R = insulin-like growth factor 1 receptor; miR-17/106b = microRNA-17/106b; MDM2 = mouse double minute 2 homolog; MMR = mismatch repair; NER = nucleotide excision repair; NOXA = Phorbol-12-myristate-13-acetate-induced protein 1; OCT-4 = octamer-binding transcription factor 4; PI3K = phosphoinositide 3-kinase; PTEN = phosphatase and tensin homolog; PUMA = p53 upregulated modulator of apoptosis; TLS = translesion synthesis.
2. Pre-Target Mechanisms
3. On-Target Mechanisms
4. Post-Target Mechanisms
5. The Role of Cellular Differentiation
6. The Role of Epigenetic Mechanisms
7. The Role of Tumor Microenvironment
8. The Role of Cancer Stem Cells
9. Summary
This entry is adapted from 10.3390/biomedicines10050972
References
- Singh, R.; Fazal, Z.; Freemantle, S.J.; Spinella, M.J. Mechanisms of cisplatin sensitivity and resistance in testicular germ cell tumors. Cancer Drug Resist. 2019, 2, 580–594.
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883.
- Chen, S.H.; Chang, J.Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136.
- Lobo, J.; Jeronimo, C.; Henrique, R. Cisplatin Resistance in Testicular Germ Cell Tumors: Current Challenges from Various Perspectives. Cancers 2020, 12, 1601.
- Holzer, A.K.; Manorek, G.H.; Howell, S.B. Contribution of the major copper influx transporter CTR1 to the cellular accumulation of cisplatin, carboplatin, and oxaliplatin. Mol. Pharmacol. 2006, 70, 1390–1394.
- Larson, C.A.; Blair, B.G.; Safaei, R.; Howell, S.B. The role of the mammalian copper transporter 1 in the cellular accumulation of platinum-based drugs. Mol. Pharmacol. 2009, 75, 324–330.
- Safaei, R.; Holzer, A.K.; Katano, K.; Samimi, G.; Howell, S.B. The role of copper transporters in the development of resistance to Pt drugs. J. Inorg. Biochem. 2004, 98, 1607–1613.
- Samimi, G.; Varki, N.M.; Wilczynski, S.; Safaei, R.; Alberts, D.S.; Howell, S.B. Increase in expression of the copper transporter ATP7A during platinum drug-based treatment is associated with poor survival in ovarian cancer patients. Clin. Cancer Res. 2003, 9, 5853–5859.
- Yang, T.; Chen, M.; Chen, T.; Thakur, A. Expression of the copper transporters hCtr1, ATP7A and ATP7B is associated with the response to chemotherapy and survival time in patients with resected non-small cell lung cancer. Oncol. Lett. 2015, 10, 2584–2590.
- Masters, J.R.; Thomas, R.; Hall, A.G.; Hogarth, L.; Matheson, E.C.; Cattan, A.R.; Lohrer, H. Sensitivity of testis tumour cells to chemotherapeutic drugs: Role of detoxifying pathways. Eur. J. Cancer 1996, 32A, 1248–1253.
- Mayer, F.; Stoop, H.; Scheffer, G.L.; Scheper, R.; Oosterhuis, J.W.; Looijenga, L.H.; Bokemeyer, C. Molecular determinants of treatment response in human germ cell tumors. Clin. Cancer Res. 2003, 9, 767–773.
- Korita, P.V.; Wakai, T.; Shirai, Y.; Matsuda, Y.; Sakata, J.; Takamura, M.; Yano, M.; Sanpei, A.; Aoyagi, Y.; Hatakeyama, K.; et al. Multidrug resistance-associated protein 2 determines the efficacy of cisplatin in patients with hepatocellular carcinoma. Oncol. Rep. 2010, 23, 965–972.
- Yamasaki, M.; Makino, T.; Masuzawa, T.; Kurokawa, Y.; Miyata, H.; Takiguchi, S.; Nakajima, K.; Fujiwara, Y.; Matsuura, N.; Mori, M.; et al. Role of multidrug resistance protein 2 (MRP2) in chemoresistance and clinical outcome in oesophageal squamous cell carcinoma. Br. J. Cancer 2011, 104, 707–713.
- Awuah, S.G.; Riddell, I.A.; Lippard, S.J. Repair shielding of platinum-DNA lesions in testicular germ cell tumors by high-mobility group box protein 4 imparts cisplatin hypersensitivity. Proc. Natl. Acad. Sci. USA 2017, 114, 950–955.
- Usanova, S.; Piee-Staffa, A.; Sied, U.; Thomale, J.; Schneider, A.; Kaina, B.; Koberle, B. Cisplatin sensitivity of testis tumour cells is due to deficiency in interstrand-crosslink repair and low ERCC1-XPF expression. Mol. Cancer 2010, 9, 248.
- Koberle, B.; Brenner, W.; Albers, A.; Usanova, S.; Thuroff, J.W.; Kaina, B. ERCC1 and XPF expression in human testicular germ cell tumors. Oncol. Rep. 2010, 23, 223–227.
- Mendoza, J.; Martinez, J.; Hernandez, C.; Perez-Montiel, D.; Castro, C.; Fabian-Morales, E.; Santibanez, M.; Gonzalez-Barrios, R.; Diaz-Chavez, J.; Andonegui, M.A.; et al. Association between ERCC1 and XPA expression and polymorphisms and the response to cisplatin in testicular germ cell tumours. Br. J. Cancer 2013, 109, 68–75.
- Cierna, Z.; Miskovska, V.; Roska, J.; Jurkovicova, D.; Pulzova, L.B.; Sestakova, Z.; Hurbanova, L.; Machalekova, K.; Chovanec, M.; Rejlekova, K.; et al. Increased levels of XPA might be the basis of cisplatin resistance in germ cell tumours. BMC Cancer 2020, 20, 17.
- Mayer, F.; Gillis, A.J.; Dinjens, W.; Oosterhuis, J.W.; Bokemeyer, C.; Looijenga, L.H. Microsatellite instability of germ cell tumors is associated with resistance to systemic treatment. Cancer Res. 2002, 62, 2758–2760.
- Velasco, A.; Riquelme, E.; Schultz, M.; Wistuba, I.I.; Villarroel, L.; Pizarro, J.; Berlin, A.; Ittmann, M.; Koh, M.S.; Leach, F.S. Mismatch repair gene expression and genetic instability in testicular germ cell tumor. Cancer Biol. Ther. 2004, 3, 977–982.
- Honecker, F.; Wermann, H.; Mayer, F.; Gillis, A.J.; Stoop, H.; van Gurp, R.J.; Oechsle, K.; Steyerberg, E.; Hartmann, J.T.; Dinjens, W.N.; et al. Microsatellite instability, mismatch repair deficiency, and BRAF mutation in treatment-resistant germ cell tumors. J. Clin. Oncol. 2009, 27, 2129–2136.
- Rudolph, C.; Melau, C.; Nielsen, J.E.; Vile Jensen, K.; Liu, D.; Pena-Diaz, J.; Rajpert-De Meyts, E.; Rasmussen, L.J.; Jorgensen, A. Involvement of the DNA mismatch repair system in cisplatin sensitivity of testicular germ cell tumours. Cell Oncol. 2017, 40, 341–355.
- Vaisman, A.; Varchenko, M.; Umar, A.; Kunkel, T.A.; Risinger, J.I.; Barrett, J.C.; Hamilton, T.C.; Chaney, S.G. The role of hMLH1, hMSH3, and hMSH6 defects in cisplatin and oxaliplatin resistance: Correlation with replicative bypass of platinum-DNA adducts. Cancer Res. 1998, 58, 3579–3585.
- Bassett, E.; Vaisman, A.; Tropea, K.A.; McCall, C.M.; Masutani, C.; Hanaoka, F.; Chaney, S.G. Frameshifts and deletions during in vitro translesion synthesis past Pt-DNA adducts by DNA polymerases beta and eta. DNA Repair 2002, 1, 1003–1016.
- Shachar, S.; Ziv, O.; Avkin, S.; Adar, S.; Wittschieben, J.; Reissner, T.; Chaney, S.; Friedberg, E.C.; Wang, Z.; Carell, T.; et al. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009, 28, 383–393.
- Ler, A.A.L.; Carty, M.P. DNA Damage Tolerance Pathways in Human Cells: A Potential Therapeutic Target. Front. Oncol. 2021, 11, 822500.
- Bagrodia, A.; Lee, B.H.; Lee, W.; Cha, E.K.; Sfakianos, J.P.; Iyer, G.; Pietzak, E.J.; Gao, S.P.; Zabor, E.C.; Ostrovnaya, I.; et al. Genetic Determinants of Cisplatin Resistance in Patients with Advanced Germ Cell Tumors. J. Clin. Oncol. 2016, 34, 4000–4007.
- Houldsworth, J.; Xiao, H.; Murty, V.V.; Chen, W.; Ray, B.; Reuter, V.E.; Bosl, G.J.; Chaganti, R.S. Human male germ cell tumor resistance to cisplatin is linked to TP53 gene mutation. Oncogene 1998, 16, 2345–2349.
- di Pietro, A.; Koster, R.; Boersma-van Eck, W.; Dam, W.A.; Mulder, N.H.; Gietema, J.A.; de Vries, E.G.; de Jong, S. Pro- and anti-apoptotic effects of p53 in cisplatin-treated human testicular cancer are cell context-dependent. Cell Cycle 2012, 11, 4552–4562.
- Di Vizio, D.; Cito, L.; Boccia, A.; Chieffi, P.; Insabato, L.; Pettinato, G.; Motti, M.L.; Schepis, F.; D’Amico, W.; Fabiani, F.; et al. Loss of the tumor suppressor gene PTEN marks the transition from intratubular germ cell neoplasias (ITGCN) to invasive germ cell tumors. Oncogene 2005, 24, 1882–1894.
- Juliachs, M.; Munoz, C.; Moutinho, C.A.; Vidal, A.; Condom, E.; Esteller, M.; Graupera, M.; Casanovas, O.; Germa, J.R.; Villanueva, A.; et al. The PDGFRbeta-AKT pathway contributes to CDDP-acquired resistance in testicular germ cell tumors. Clin. Cancer Res. 2014, 20, 658–667.
- Koster, R.; di Pietro, A.; Timmer-Bosscha, H.; Gibcus, J.H.; van den Berg, A.; Suurmeijer, A.J.; Bischoff, R.; Gietema, J.A.; de Jong, S. Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J. Clin. Investig. 2010, 120, 3594–3605.
- Feldman, D.R.; Iyer, G.; Van Alstine, L.; Patil, S.; Al-Ahmadie, H.; Reuter, V.E.; Bosl, G.J.; Chaganti, R.S.; Solit, D.B. Presence of somatic mutations within PIK3CA, AKT, RAS, and FGFR3 but not BRAF in cisplatin-resistant germ cell tumors. Clin. Cancer Res. 2014, 20, 3712–3720.
- Selfe, J.; Goddard, N.C.; McIntyre, A.; Taylor, K.R.; Renshaw, J.; Popov, S.D.; Thway, K.; Summersgill, B.; Huddart, R.A.; Gilbert, D.C.; et al. IGF1R signalling in testicular germ cell tumour cells impacts on cell survival and acquired cisplatin resistance. J. Pathol. 2018, 244, 242–253.
- Noel, E.E.; Yeste-Velasco, M.; Mao, X.; Perry, J.; Kudahetti, S.C.; Li, N.F.; Sharp, S.; Chaplin, T.; Xue, L.; McIntyre, A.; et al. The association of CCND1 overexpression and cisplatin resistance in testicular germ cell tumors and other cancers. Am. J. Pathol. 2010, 176, 2607–2615.
- Timmer-Bosscha, H.; de Vries, E.G.; Meijer, C.; Oosterhuis, J.W.; Mulder, N.H. Differential effects of all-trans-retinoic acid, docosahexaenoic acid, and hexadecylphosphocholine on cisplatin-induced cytotoxicity and apoptosis in a cisplantin-sensitive and resistant human embryonal carcinoma cell line. Cancer Chemother. Pharmacol. 1998, 41, 469–476.
- Skotheim, R.I.; Lind, G.E.; Monni, O.; Nesland, J.M.; Abeler, V.M.; Fossa, S.D.; Duale, N.; Brunborg, G.; Kallioniemi, O.; Andrews, P.W.; et al. Differentiation of human embryonal carcinomas in vitro and in vivo reveals expression profiles relevant to normal development. Cancer Res. 2005, 65, 5588–5598.
- Honecker, F.; Rohlfing, T.; Harder, S.; Braig, M.; Gillis, A.J.; Glaesener, S.; Barett, C.; Bokemeyer, C.; Buck, F.; Brummendorf, T.H.; et al. Proteome analysis of the effects of all-trans retinoic acid on human germ cell tumor cell lines. J. Proteom. 2014, 96, 300–313.
- Nettersheim, D.; Gillis, A.; Biermann, K.; Looijenga, L.H.; Schorle, H. The seminoma cell line TCam-2 is sensitive to HDAC inhibitor depsipeptide but tolerates various other chemotherapeutic drugs and loss of NANOG expression. Genes Chromosomes Cancer 2011, 50, 1033–1042.
- Nettersheim, D.; Arndt, I.; Sharma, R.; Riesenberg, S.; Jostes, S.; Schneider, S.; Holzel, M.; Kristiansen, G.; Schorle, H. The cancer/testis-antigen PRAME supports the pluripotency network and represses somatic and germ cell differentiation programs in seminomas. Br. J. Cancer 2016, 115, 454–464.
- Gutekunst, M.; Oren, M.; Weilbacher, A.; Dengler, M.A.; Markwardt, C.; Thomale, J.; Aulitzky, W.E.; van der Kuip, H. p53 hypersensitivity is the predominant mechanism of the unique responsiveness of testicular germ cell tumor (TGCT) cells to cisplatin. PLoS ONE 2011, 6, e19198.
- Gutekunst, M.; Mueller, T.; Weilbacher, A.; Dengler, M.A.; Bedke, J.; Kruck, S.; Oren, M.; Aulitzky, W.E.; van der Kuip, H. Cisplatin hypersensitivity of testicular germ cell tumors is determined by high constitutive Noxa levels mediated by Oct-4. Cancer Res. 2013, 73, 1460–1469.
- Mueller, T.; Mueller, L.P.; Luetzkendorf, J.; Voigt, W.; Simon, H.; Schmoll, H.J. Loss of Oct-3/4 expression in embryonal carcinoma cells is associated with induction of cisplatin resistance. Tumour. Biol. 2006, 27, 71–83.
- Wu, Y.C.; Ling, T.Y.; Lu, S.H.; Kuo, H.C.; Ho, H.N.; Yeh, S.D.; Shen, C.N.; Huang, Y.H. Chemotherapeutic sensitivity of testicular germ cell tumors under hypoxic conditions is negatively regulated by SENP1-controlled sumoylation of OCT4. Cancer Res. 2012, 72, 4963–4973.
- Abada, P.B.; Howell, S.B. Cisplatin induces resistance by triggering differentiation of testicular embryonal carcinoma cells. PLoS ONE 2014, 9, e87444.
- Pierpont, T.M.; Lyndaker, A.M.; Anderson, C.M.; Jin, Q.; Moore, E.S.; Roden, J.L.; Braxton, A.; Bagepalli, L.; Kataria, N.; Hu, H.Z.; et al. Chemotherapy-Induced Depletion of OCT4-Positive Cancer Stem Cells in a Mouse Model of Malignant Testicular Cancer. Cell Rep. 2017, 21, 1896–1909.
- Taylor-Weiner, A.; Zack, T.; O’Donnell, E.; Guerriero, J.L.; Bernard, B.; Reddy, A.; Han, G.C.; AlDubayan, S.; Amin-Mansour, A.; Schumacher, S.E.; et al. Genomic evolution and chemoresistance in germ-cell tumours. Nature 2016, 540, 114–118.
- Mueller, T.; Mueller, L.P.; Holzhausen, H.J.; Witthuhn, R.; Albers, P.; Schmoll, H.J. Histological evidence for the existence of germ cell tumor cells showing embryonal carcinoma morphology but lacking OCT4 expression and cisplatin sensitivity. Histochem. Cell Biol. 2010, 134, 197–204.
- Looijenga, L.H.; Stoop, H.; de Leeuw, H.P.; de Gouveia Brazao, C.A.; Gillis, A.J.; van Roozendaal, K.E.; van Zoelen, E.J.; Weber, R.F.; Wolffenbuttel, K.P.; van Dekken, H.; et al. POU5F1 (OCT3/4) identifies cells with pluripotent potential in human germ cell tumors. Cancer Res. 2003, 63, 2244–2250.
- Koster, R.; van Vugt, M.A.; Timmer-Bosscha, H.; Gietema, J.A.; de Jong, S. Unravelling mechanisms of cisplatin sensitivity and resistance in testicular cancer. Expert Rev. Mol. Med. 2013, 15, e12.
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated Molecular Characterization of Testicular Germ Cell Tumors. Cell Rep. 2018, 23, 3392–3406.
- Lafin, J.T.; Bagrodia, A.; Woldu, S.; Amatruda, J.F. New insights into germ cell tumor genomics. Andrology 2019, 7, 507–515.
- Young, J.C.; Kerr, G.; Micati, D.; Nielsen, J.E.; Rajpert-De Meyts, E.; Abud, H.E.; Loveland, K.L. WNT signalling in the normal human adult testis and in male germ cell neoplasms. Hum. Reprod 2020, 35, 1991–2003.
- ten Berge, D.; Kurek, D.; Blauwkamp, T.; Koole, W.; Maas, A.; Eroglu, E.; Siu, R.K.; Nusse, R. Embryonic stem cells require Wnt proteins to prevent differentiation to epiblast stem cells. Nat. Cell Biol. 2011, 13, 1070–1075.
- Bayerl, J.; Ayyash, M.; Shani, T.; Manor, Y.S.; Gafni, O.; Massarwa, R.; Kalma, Y.; Aguilera-Castrejon, A.; Zerbib, M.; Amir, H.; et al. Principles of signaling pathway modulation for enhancing human naive pluripotency induction. Cell Stem. Cell 2021, 28, 1549–1565.
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84.
- Chovanec, M.; Cierna, Z.; Miskovska, V.; Machalekova, K.; Kalavska, K.; Rejlekova, K.; Svetlovska, D.; Macak, D.; Spanik, S.; Kajo, K.; et al. betacatenin is a marker of poor clinical characteristics and suppressed immune infiltration in testicular germ cell tumors. BMC Cancer 2018, 18, 1062.
- Schmidtova, S.; Kalavska, K.; Liskova, V.; Plava, J.; Miklikova, S.; Kucerova, L.; Matuskova, M.; Rojikova, L.; Cierna, Z.; Rogozea, A.; et al. Targeting of Deregulated Wnt/beta-Catenin Signaling by PRI-724 and LGK974 Inhibitors in Germ Cell Tumor Cell Lines. Int. J. Mol. Sci. 2021, 22, 4263.
- Buljubasic, R.; Buljubasic, M.; Bojanac, A.K.; Ulamec, M.; Vlahovic, M.; Jezek, D.; Bulic-Jakus, F.; Sincic, N. Epigenetics and testicular germ cell tumors. Gene 2018, 661, 22–33.
- Facchini, G.; Rossetti, S.; Cavaliere, C.; D’Aniello, C.; Di Franco, R.; Iovane, G.; Grimaldi, G.; Piscitelli, R.; Muto, P.; Botti, G.; et al. Exploring the molecular aspects associated with testicular germ cell tumors: A review. Oncotarget 2018, 9, 1365–1379.
- Sonnenburg, D.; Spinella, M.J.; Albany, C. Epigenetic Targeting of Platinum Resistant Testicular Cancer. Curr. Cancer Drug Targets 2016, 16, 789–795.
- Fazal, Z.; Singh, R.; Fang, F.; Bikorimana, E.; Baldwin, H.; Corbet, A.; Tomlin, M.; Yerby, C.; Adra, N.; Albany, C.; et al. Hypermethylation and global remodelling of DNA methylation is associated with acquired cisplatin resistance in testicular germ cell tumours. Epigenetics 2021, 16, 1071–1084.
- Wermann, H.; Stoop, H.; Gillis, A.J.; Honecker, F.; van Gurp, R.J.; Ammerpohl, O.; Richter, J.; Oosterhuis, J.W.; Bokemeyer, C.; Looijenga, L.H. Global DNA methylation in fetal human germ cells and germ cell tumours: Association with differentiation and cisplatin resistance. J. Pathol. 2010, 221, 433–442.
- Koul, S.; McKiernan, J.M.; Narayan, G.; Houldsworth, J.; Bacik, J.; Dobrzynski, D.L.; Assaad, A.M.; Mansukhani, M.; Reuter, V.E.; Bosl, G.J.; et al. Role of promoter hypermethylation in Cisplatin treatment response of male germ cell tumors. Mol. Cancer 2004, 3, 16.
- Martinelli, C.; Lengert, A.V.H.; Carcano, F.M.; Silva, E.C.A.; Brait, M.; Lopes, L.F.; Vidal, D.O. MGMT and CALCA promoter methylation are associated with poor prognosis in testicular germ cell tumor patients. Oncotarget 2017, 8, 50608–50617.
- Acunzo, M.; Romano, G.; Wernicke, D.; Croce, C.M. MicroRNA and cancer—A brief overview. Adv. Biol. Regul. 2015, 57, 1–9.
- Das, M.K.; Haugen, O.P.; Haugen, T.B. Diverse Roles and Targets of miRNA in the Pathogenesis of Testicular Germ Cell Tumour. Cancers 2022, 14, 1190.
- Port, M.; Glaesener, S.; Ruf, C.; Riecke, A.; Bokemeyer, C.; Meineke, V.; Honecker, F.; Abend, M. Micro-RNA expression in cisplatin resistant germ cell tumor cell lines. Mol. Cancer 2011, 10, 52.
- Kalavska, K.; Kucerova, L.; Schmidtova, S.; Chovanec, M.; Mego, M. Cancer Stem Cell Niche and Immune-Active Tumor Microenvironment in Testicular Germ Cell Tumors. Adv. Exp. Med. Biol. 2020, 1226, 111–121.
- Loveland, K.L.; Klein, B.; Pueschl, D.; Indumathy, S.; Bergmann, M.; Loveland, B.E.; Hedger, M.P.; Schuppe, H.C. Cytokines in Male Fertility and Reproductive Pathologies: Immunoregulation and Beyond. Front. Endocrinol. 2017, 8, 307.
- Siska, P.J.; Johnpulle, R.A.N.; Zhou, A.; Bordeaux, J.; Kim, J.Y.; Dabbas, B.; Dakappagari, N.; Rathmell, J.C.; Rathmell, W.K.; Morgans, A.K.; et al. Deep exploration of the immune infiltrate and outcome prediction in testicular cancer by quantitative multiplexed immunohistochemistry and gene expression profiling. Oncoimmunology 2017, 6, e1305535.
- Lama-Sherpa, T.D.; Shevde, L.A. An Emerging Regulatory Role for the Tumor Microenvironment in the DNA Damage Response to Double-Strand Breaks. Mol. Cancer Res. 2020, 18, 185–193.
- Kalavska, K.; Sestakova, Z.; Mlcakova, A.; Kozics, K.; Gronesova, P.; Hurbanova, L.; Miskovska, V.; Rejlekova, K.; Svetlovska, D.; Sycova-Mila, Z.; et al. Are Changes in the Percentage of Specific Leukocyte Subpopulations Associated with Endogenous DNA Damage Levels in Testicular Cancer Patients? Int. J. Mol. Sci. 2021, 22, 8281.
- Cierna, Z.; Mego, M.; Miskovska, V.; Machalekova, K.; Chovanec, M.; Svetlovska, D.; Hainova, K.; Rejlekova, K.; Macak, D.; Spanik, S.; et al. Prognostic value of programmed-death-1 receptor (PD-1) and its ligand 1 (PD-L1) in testicular germ cell tumors. Ann. Oncol. 2016, 27, 300–305.
- Chovanec, M.; Cierna, Z.; Miskovska, V.; Machalekova, K.; Svetlovska, D.; Kalavska, K.; Rejlekova, K.; Spanik, S.; Kajo, K.; Babal, P.; et al. Prognostic role of programmed-death ligand 1 (PD-L1) expressing tumor infiltrating lymphocytes in testicular germ cell tumors. Oncotarget 2017, 8, 21794–21805.
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472.
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem. Cell 2015, 16, 225–238.
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell Physiol. 2019, 234, 8381–8395.
- Dianat-Moghadam, H.; Heidarifard, M.; Jahanban-Esfahlan, R.; Panahi, Y.; Hamishehkar, H.; Pouremamali, F.; Rahbarghazi, R.; Nouri, M. Cancer stem cells-emanated therapy resistance: Implications for liposomal drug delivery systems. J. Control Release 2018, 288, 62–83.
- Bai, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Cancer stem cell in breast cancer therapeutic resistance. Cancer Treat Rev. 2018, 69, 152–163.
- Xu, S.L.; Zeng, D.Z.; Dong, W.G.; Ding, Y.Q.; Rao, J.; Duan, J.J.; Liu, Q.; Yang, J.; Zhan, N.; Liu, Y.; et al. Distinct patterns of ALDH1A1 expression predict metastasis and poor outcome of colorectal carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 2976–2986.
- Liu, Y.; Lv, D.L.; Duan, J.J.; Xu, S.L.; Zhang, J.F.; Yang, X.J.; Zhang, X.; Cui, Y.H.; Bian, X.W.; Yu, S.C. ALDH1A1 expression correlates with clinicopathologic features and poor prognosis of breast cancer patients: A systematic review and meta-analysis. BMC Cancer 2014, 14, 444.
- Li, X.; Wan, L.; Geng, J.; Wu, C.L.; Bai, X. Aldehyde dehydrogenase 1A1 possesses stem-like properties and predicts lung cancer patient outcome. J. Thorac. Oncol. 2012, 7, 1235–1245.
- Nishikawa, S.; Konno, M.; Hamabe, A.; Hasegawa, S.; Kano, Y.; Ohta, K.; Fukusumi, T.; Sakai, D.; Kudo, T.; Haraguchi, N.; et al. Aldehyde dehydrogenase high gastric cancer stem cells are resistant to chemotherapy. Int. J. Oncol. 2013, 42, 1437–1442.
- Kang, E.J.; Jung, H.; Woo, O.H.; Park, K.H.; Woo, S.U.; Yang, D.S.; Kim, A.R.; Lee, J.B.; Kim, Y.H.; Kim, J.S.; et al. Association of aldehyde dehydrogenase 1 expression and biologically aggressive features in breast cancer. Neoplasma 2014, 61, 352–362.
- Schmidtova, S.; Kalavska, K.; Gercakova, K.; Cierna, Z.; Miklikova, S.; Smolkova, B.; Buocikova, V.; Miskovska, V.; Durinikova, E.; Burikova, M.; et al. Disulfiram Overcomes Cisplatin Resistance in Human Embryonal Carcinoma Cells. Cancers 2019, 11, 1224.
More