・ Degenerative retina in RP is exposed to high-level O2 and thereby damaged by ROS.
・ Microglia as well as photoreceptor cells are injured by ROS.
・ Oxidative microglial activation promotes microgliosis and photoreceptor cell death in RP.
・ Oxidative DNA damage mediates MUTYH-SSBs-PARP signaling to induce microglial activation.
- Oxidative DNA damage
- Retinitis Pigmentosa
1. Introduction
The cellular organelles and molecules of the human body are always at risk of being oxidized by reactive oxygen species (ROS), which are produced as a regular part of the body’s activity. ROS such as superoxide (1O2), hydrogen peroxide (H2O2), and hydroxyl radicals (•OH) are generated in the process of oxidative phosphorylation and ATP synthesis in the mitochondria [1]. Alternatively, ROS can be produced by NADPH oxidases (NOXs), which are transmembrane enzymes that form a 1O2-producing protein complex upon activation [2]. NOXs are critical for the body’s immune defense against infected bacteria as well as for health and fighting disease in a variety of tissues including the retina.
To protect the organelles and molecules against ROS, the cells have an elaborate defense system to neutralize or catalyze ROS and repair ROS-induced damage (Figure 1). For example, superoxide dismutase (SOD) catalyzes the dismutation of 1O2 into oxygen (O2) and H2O2. Catalase breaks down •OH into O2 and water (H2O). Glutathione peroxidase (GPx) catalyzes H2O2 into H2O, with the conversion of glutathione (GSH) to its oxidized disulfide form (GSSG) [3].
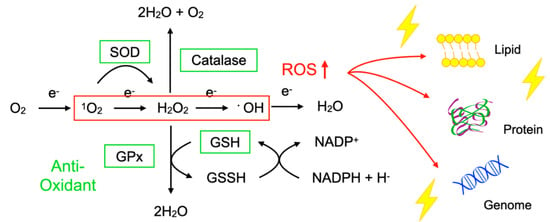
Figure 1. Imbalance between reactive oxygen species (ROS) and anti-oxidants impairs the function of macromolecules. ROS including superoxide (1O2), hydrogen peroxide (H2O2), and hydroxyl radicals (•OH) are generated during oxidative phosphorylation in the mitochondria. Superoxide dismutase (SOD) catalyzes the 1O2 into oxygen (O2) and H2O2. Catalase breaks down •OH into O2 and water (H2O). Glutathione peroxidase (GPx) catalyzes H2O2 into H2O, with the conversion of glutathione (GSH) to its oxidized disulfide form (GSSG). An imbalance between ROS production (red box) and anti-oxidant capacity (green box) results in the accumulation of oxidative insults (lightning symbol) to the cellular lipids, proteins, and nucleic acids. ↑, increase.
Oxidative stress is a state in which the balance between the production of ROS and the anti-oxidant defense system is impaired. The production of ROS is markedly increased in various disease conditions including inflammation, metabolic dysfunction, cancer, and neurodegeneration. Excessive ROS insults the cellular macromolecules such as nucleic acids, proteins, and lipids (Figure 1), leading to cellular dysfunction, transdifferentiation, or death. Accordingly, oxidative stress is generally deemed to be detrimental to human health; however, it should be noted that, in some conditions, ROS are required to mediate the body’s protection against infection and tissue injury [4].
2. Retinitis Pigmentosa and Oxidative Stress
2.1. Etiology of Retinitis Pigmentosa
Retinitis pigmentosa (RP) comprises a group of inherited retinal degeneration states that, without effective treatment, lead to blindness [5][6][5,6]. Genetic mutations associated with RP have been identified in more than 90 genes, most of which are related to the function and maintenance of rod photoreceptor cells. Rod cells are responsible for vision in dim light, and the symptoms of RP typically start with night blindness due to the dysfunction and death of rod cells. Ring scotoma at the mid-peripheral retina, which corresponds to the region containing the highest rod density, is also associated with RP. At this early stage of the disease, the daily lives and activity levels of patients are usually not severely affected. However, following rod cell loss, the remaining cone photoreceptor cells are gradually but progressively impaired, leading to constriction of the visual field and eventually the loss of central vision. This impairment of cone-mediated daylight vision is the most debilitating aspect of RP. Based on these clinical features, RP is also categorized as rod-cone dystrophy [7].
As can be seen from the variety of causal genes, RP patients show diverse heterogeneity in their phenotype. For example, the onset age of night blindness or other symptoms ranges from early childhood to the mid-30s or even later. The central visual function and its progression also have significant inter-individual variability among patients with mutations in the same gene or even among family members with the same mutation. Recent advances in genotype–phenotype technology have led to a better characterization of each causal gene in RP, but there are still significant gaps between genetic results and clinical findings [8].
Despite the heterogeneity in disease progression, there are shared clinical characteristics in RP, including the appearance of the fundus. It has been suggested that some disease-modifying factors may underlie the disease as a common etiology of RP. For example, following the rod cell loss, the degenerative retina, which largely reduces the O2 consumption, is exposed to higher levels of O2 and thereby damaged by ROS, suggesting that oxidative stress may promote rod and cone degeneration in RP [9]. Other biological factors such as inflammation, autophagy, and metabolic dysfunction have been suggested to modify the disease progression of RP [10][11][12][10,11,12]. Elucidation of the biological mechanisms underlying the rod and cone degeneration in RP will be important for gaining a better understanding of the disease and for the development of novel therapies targeting specific biological pathways.
2.2. Oxidative Damage in Animal Models of RP
2.2.1. Evidence of Increased Oxidative Damage in the RP Retina
Increased ROS in the tissue can be experimentally detected by probes such as dihydroethidium (DHE) and CellROX™, which react with ROS to produce a fluorescent signal. After an in vivo administration of DHE, the red fluorescent signal can be detected in the outer retina of rd1 mice, a murine model of RP that have a mutation in Pde6b and a confounding mutation in Gpr179 [13][14][13,14]. This ROS signal is not observed in the normal retina, indicating that the production of ROS is markedly increased in the degenerative loci of RP.
Oxidized lipids, proteins, and nucleic acids can also be visualized by the specific antibodies against the oxidized residue of each molecule. For example, malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE) are byproducts of lipid peroxidation; protein carbonyls and nitrotyrosine are markers of protein oxidation; and 8-oxo-7,8-dihydroguannine (8-oxoguanine, or 8-oxoG) is a major form of oxidized nucleic acids. In several models of RP, it was shown that oxidized lipids, proteins, and nucleic acids are substantially increased in the degenerative retina, especially in the photoreceptor layer (Figure 2A,B) [15][16][15,16].
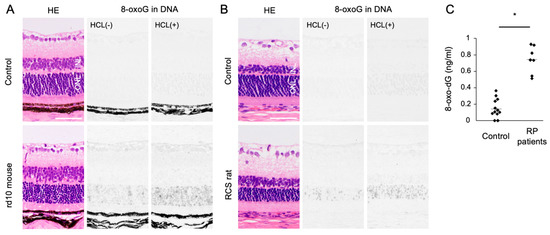
Figure 2. The Figure is reproduced from [16] [16] with copyright permission. Accumulation of oxidative DNA damage in retinitis pigmentosa (RP). (A,B) Hematoxylin and eosin (HE) staining and immunohistochemical staining of 8-oxo-7,8-dihydroguannine (8-oxoG) in the retina of rd10 mice (A) and Royal College of Surgeons (RCS) rats (B), two genetically different models of retinitis pigmentosa. Per staining, HCl pretreatment was used to denature the nuclear DNA, thereby enhancing the detection of 8-oxoG in the nuclear DNA. Note that 8-oxo-G accumulation is substantially increased in the outer nuclear layer (ONL) of RP mice. Scale bar, 50 μm. INL: inner nuclear layer. (C) Enzyme-linked immunosorbent assay for 8-oxo-dG in the vitreous of RP patients and controls (patients with idiopathic epiretinal membrane). The vitreous levels of 8-oxo-dG are increased in RP patients. * p = 0.0003
In addition to the photoreceptor cells, immune cells such as microglial cells or macrophages extensively infiltrate these outer retinal regions and express oxidative molecules such as NOX2, suggesting that these inflammatory cells may be an alternative source of ROS in the RP retina [13].
2.2.2. Role of Oxidative Damage in RP
Although oxidative stress has bidirectional roles (alternately beneficial and detrimental), laboratory findings suggest that oxidative stress acts to promote the retinal degeneration in RP. Yamada et al. showed that mice placed in 75% O2 for 2 weeks exhibited significant degeneration of photoreceptor cells, supporting the notion that a higher O2 supply may be deleterious to the retina [17].
In rd1 mice, Komeima et al. tested a cocktail of four anti-oxidants (vitamin E, a SOD mimetic, vitamin C, and a-lipoic acid), and they demonstrated that this anti-oxidant treatment substantially reduced the accumulation of oxidized lipids and protected cone cells against death. rd10 mice with a Pde6b mutation are another model of RP, and exhibit a slower progression of retinal degeneration [18]. Lee et al. treated rd10 mice with oral N-acetylcysteine (NAC), which replenishes intracellular GSH, and found that NAC substantially protected rod and cone photoreceptor cells [19]. We also assessed oral NAC for rd10 mice, and we found that NAC exerted neuroprotective as well as immunosuppressive effects [20]. The therapeutic effects of anti-oxidants have been observed in several animal models with different genetic mutations, suggesting that oxidative stress may be a common important pathology of the retinal degeneration in RP.
One limitation of these laboratory studies is that, in most of the experiments, the dosage of anti-oxidant drugs was far higher than that used in human medicine or dietary supplements. In addition, the treatment was often initiated at the very early phase in experimental models. Given that RP patients are usually referred to a hospital at the mid-phase or even late phase of the disease, the efficacy of a treatment in experimental models should be carefully interpreted.
2.2.3. Modification of Anti-Oxidant Genes in RP
Gene modification to enhance the body’s anti-oxidant capacity is another approach to combat oxidative stress. Usui et al. showed that transgenic overexpressions of SOD1 and GPx4, which catalyze 1O2 and H2O2, delay the cone degeneration in rd1 mice [21]. Although transgenic overexpression is not applicable to humans, Xiong et al. demonstrated that the viral vector-mediated retinal gene transfer of an anti-oxidant gene had therapeutic potential in RP models. In rd1, rd10, and Rhodopsin−/− mice, the adeno-associated virus (AAV) vector-mediated delivery of nuclear factor erythroid-derived 2-like 2 (NRF2), a transcription factor that boosts detoxifying and anti-oxidant genes on oxidative stimulation, is effective for cone survival [22]. Because gene therapy using AAV vector has been approved for Leber’s congenital amaurosis and has been widely tested in clinical trials for inherited retinal degeneration and other retinal diseases, local and long-lasting anti-oxidant therapy may be an alternative strategy for chronic retinal degenerative disorders including RP [23][24][23,24].
2.3. Clinical Evidence of Oxidative Stress in RP
Since human retinal samples are rarely obtained with an immediate sample preparation to prevent post-mortem oxidation, oxidative stress in RP patients has been analyzed using the aqueous humor, vitreous body, and peripheral blood samples. In ocular samples, we showed that 8-oxo-7,8-dyhydro-2′-deoxyguanosine (8-oxo-dG), a marker of oxidative DNA damage, is increased more than 5-fold in the vitreous of RP patients compared to controls without retinal degeneration (Figure 2C) [16]. Consistent with this finding, Campochiaro et al. demonstrated an approximately 2-fold increase of protein carbonyl contents in the aqueous humor of RP patients [25]. On the other hand, anti-oxidant molecules such as GSH and SOD3 were decreased in the aqueous humor of RP patients [26]. These findings suggest that an oxidative imbalance occurs in the eyes of RP patients, which is consistent with findings in experimental models.
The oxidant and anti-oxidant profiles in the peripheral blood of RP patients have shown some conflicting results. Martínez-Fernández de la Cámara et al. reported increased oxidative markers (e.g., nitrotyrosine and thiobarbituric acid reactive substances) but decreased anti-oxidant SOD3 in RP patients [26]. In contrast, Campochiaro et al. demonstrated no differences in protein carbonyls, the GSH/GSSH ratio, or SOD3 in the serum of RP patients [25]. In our cohort of 52 RP patients, we investigated an oxidant marker (hexanoyl-lysine (HEL)) and three anti-oxidant markers (SOD3, GPx, and potential anti-oxidant (PAO)). We observed no significant difference in these four markers between RP patients and healthy controls; however, a subgroup analysis showed that the serum SOD3 activity was significantly lower in the RP patients with severe degeneration involving the macula [27]. In addition, the lower serum SOD3 activity in the RP patients was related to worse visual acuity and macular retinal sensitivity [27]. These data suggest that the decline of serum SOD3 activity is associated with the loss of cone-mediated central vision.
There are two possible interpretations for the lower serum SOD3 in patients with advanced RP. One possibility is that the serum SOD3 level falls in response to retinal degeneration and increased ocular ROS in RP. Another possibility is that RP patients with a lower baseline anti-oxidant capacity have a faster disease progression. This point should be addressed in future studies by directly comparing oxidant/anti-oxidant molecules between aqueous and serum samples as well as by following up the patients to determine the longitudinal changes of the serum SOD3 activity and central vision.